






基础
GEO表达矩阵的来源
- 不分方向
- GEO (常用)
- NHANES (临床数据)
- 肿瘤专属
- TCGA (常用)
- ICGC
- CCLE
- SEER (临床数据)
有了表达矩阵,如何做后续分析?
- 数据下载
- 差异分析(芯片和转录组不同)
- WGCNA(加权共表达网络) (差异分析的替代方案,如果样本数>15,且分组较为杂乱可以使用)
- 富集分析
- ORA
- GSEA
- 蛋白-蛋白互作网络(PPI)
常见图表
- 差异表达热图:
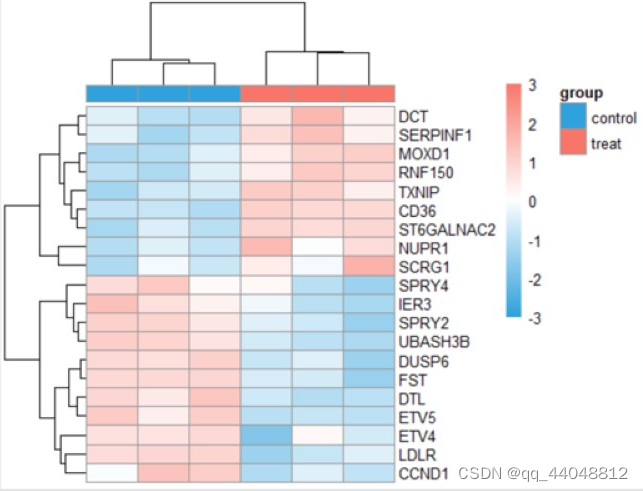
- 箱线图:

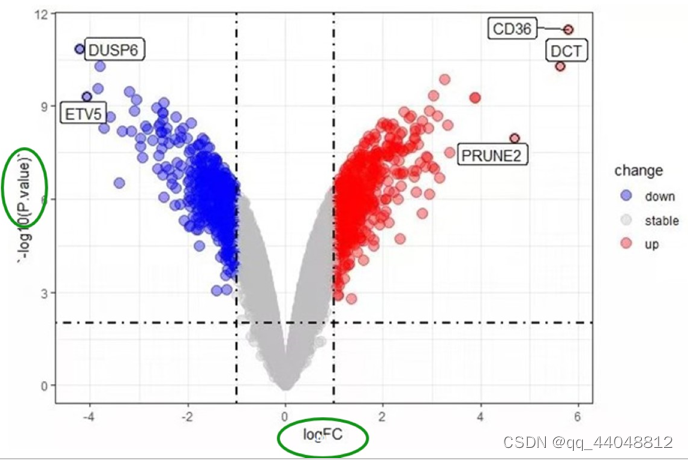
- 火山图:
- PCA图:
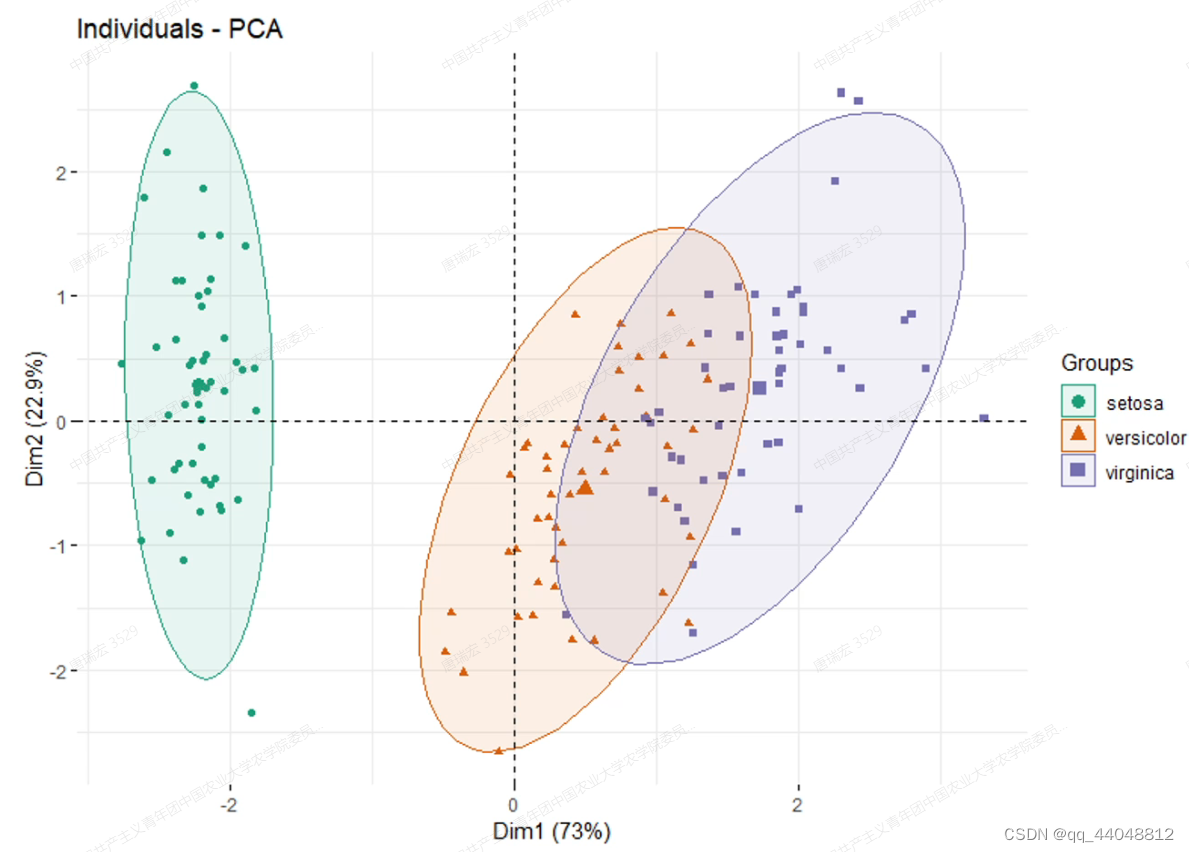
- Dim1和2的"能解释…%的变异"不做要求。
- 看同一分组是否聚成一簇(组内重复好);中心点之间是否有距离(组间差异大)。
- 通过查看PCA图,可以分辨离群点(离得太远了);查看组内是否有亚组(同组样本分为多簇)。
- 注意,PCA点(样品数)比较多时,代码默认在样本周围生成椭圆,椭圆中心点比普通样本点大一圈。
作图数据
log2FC:- Foldchange (FC):处理组平均值 / 对照组平均值
- log2Foldchange (logFC):
∑
处理组样品
log
2
(
表达量
+
1
)
处理组样品数
−
∑
对照组样品
log
2
(
表达量
+
1
)
对照组样品数
\frac{\sum^{处理组样品}\log_2(表达量+1)}{处理组样品数} - \frac{\sum^{对照组样品}\log_2(表达量+1)}{对照组样品数}
处理组样品数∑处理组样品log2(表达量+1)−对照组样品数∑对照组样品log2(表达量+1)
- 这里的表达量是未经过log2转换的;对于转换后的表达量,仅需求平均数即可。
表达量+1,以避免非正数。
- logFC应该<10。
- logFC常见阈值:1、2、1.2、1.5、log2(1.5)(当原始表达量相差1.5倍时,就认为有差异)……均可,主要看阈值筛选出的差异基因是否足够支撑富集分析。
p-value:使用原始P值和校正后P值均可,还是看差异基因是否足够多。
GEO背景知识和表达芯片分析流程

- 思路:差异处理或材料 → 差异基因 → 功能富集 → 解释处理或不同的材料是如何导致表型变化的 → 研究关注通路
基础
- 一个Series(实验设计, GSE…)里面包含了Platforms(GPL…)和Sample(GSM…)。
- 从GSM中可以看到作者提供的原始文件和原始或经处理的数据。
- 对于基因芯片,平台即为基因芯片的编号,可以由探针编号得到
- 由于探针是针对序列设计的,并不代表基因,因此以前的基因芯片可能由于基因数据库更新,造成基因芯片探针与基因的一一对应关系被破坏。需要将无对应基因的探针删除。
基因芯片表达矩阵
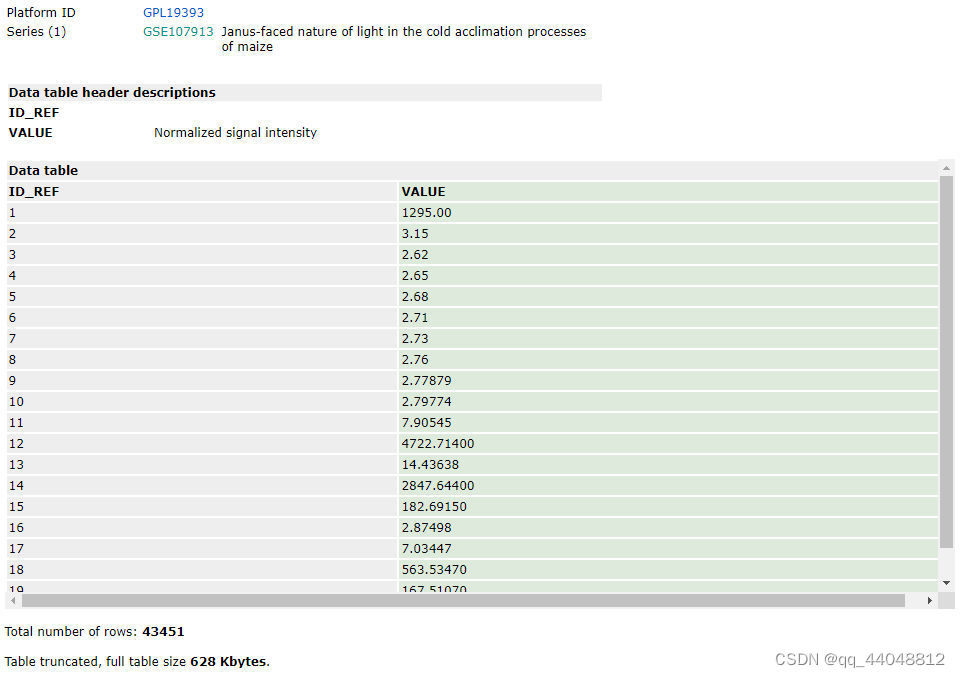
ID_REF:基因芯片的探针ID,可以通过Platform与基因对应。VALUE:得到的原始数据或经过处理后(该处为归一化)的数据。- 数据能否直接作为表达矩阵使用?
- 文件大小应>500 kb,如果很小,可能是空文件。
GEO 处理流程
理论
-
找数据,找到GSE编号
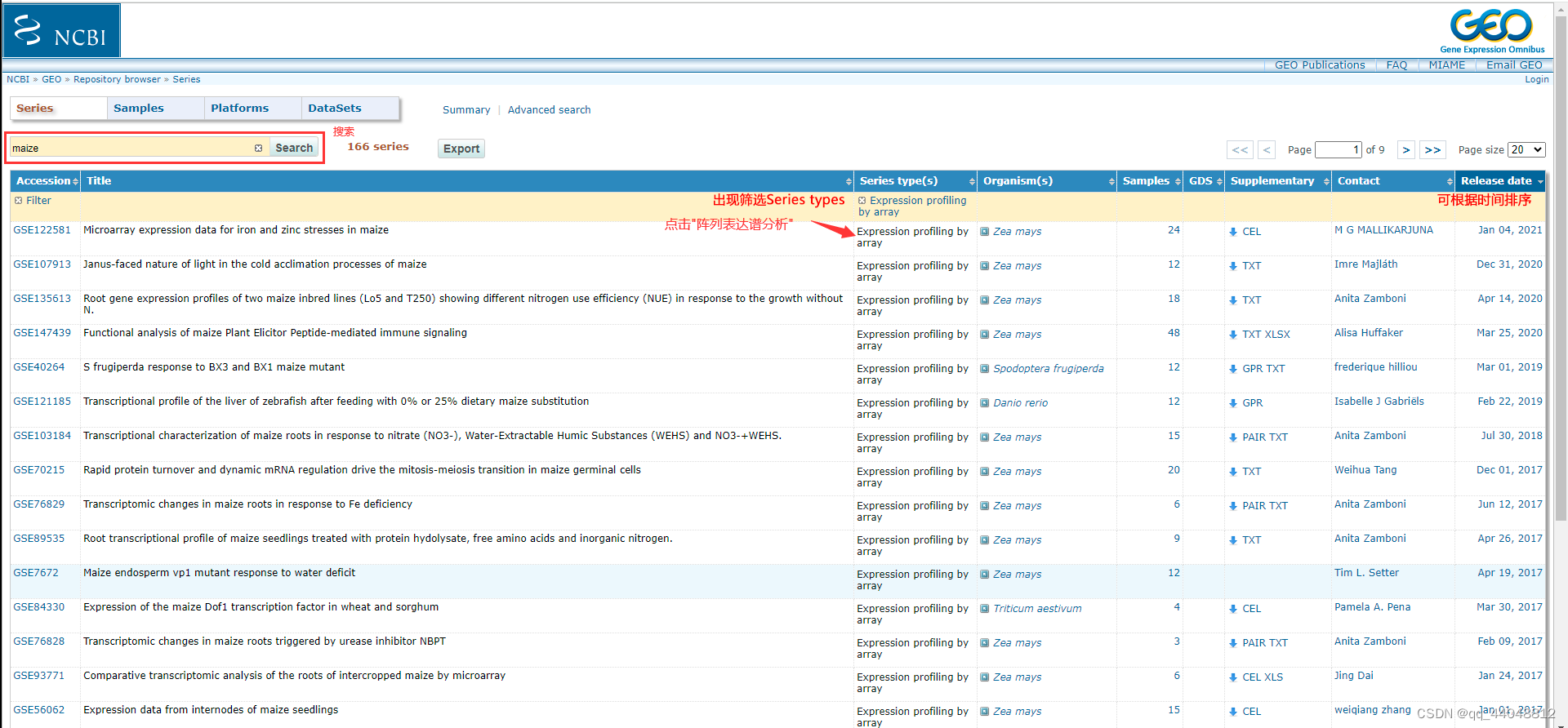
- 因为是在做数据挖掘,不要选样本数太少的GSE(2-4个)的数据(对于医学领域是,但是不知道对于植物(玉米)是不是这样)。
-
下载数据
- 表达矩阵
- 分组信息
- GPL编号(探针注释)
-
数据探索
- 查看分组之间是否有差异和表达模式:PCA、热图(取方差最大的1000个基因)。
-
差异分析及可视化
- 得到P-value,logFC。
- 画火山图、差异基因热图。
- 热图只需要画部分基因的,全部画看不出来差别。
-
富集分析(KEGG、GO)
- 富集分析除了看全局差异基因的富集以外,还可以看现有的差异基因与其他热点基因集或预测靶基因的交集。
如何看作者提供的表达矩阵是否能直接用来差异分析?
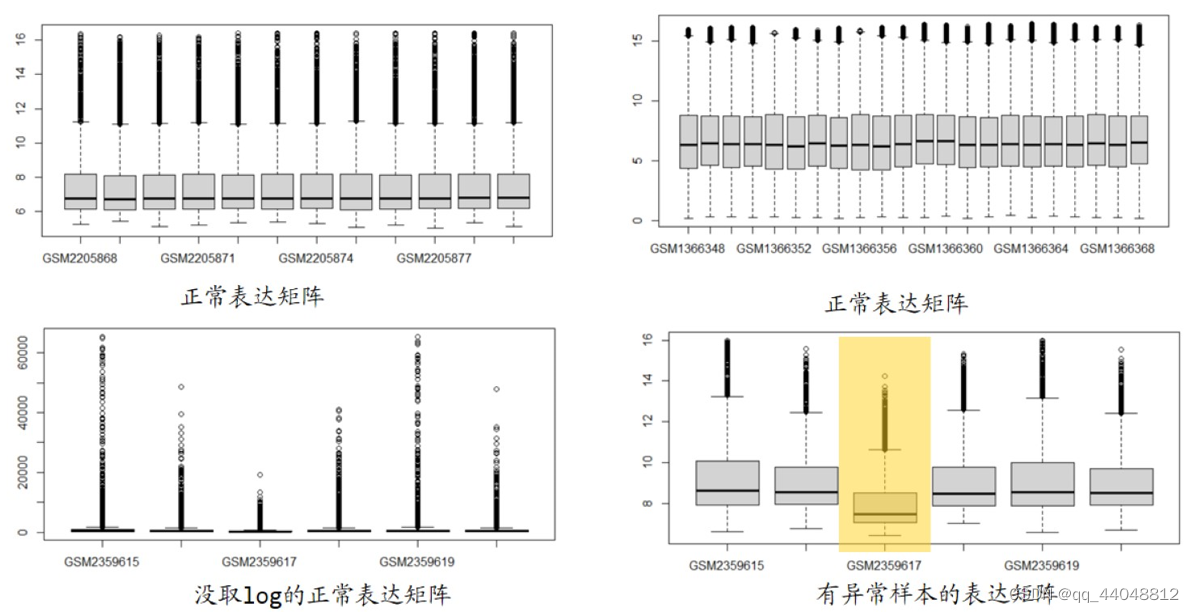
- 表达矩阵取过log2了吗?查看数据范围(分布)
- 芯片取log2后的值大多在0-20间,最高不超过24。如果最大值高达几万,肯定没有经过log2。
- 当最大值几万,还有负值时,认为数据是错误的。
- 芯片差异分析的起点是一个取过log2的表达矩阵,需要查看是否已被log2转换,或者是否被二次log2转换。
- 表达矩阵中的负值多吗?
- 如果负值正值一半一半,则数据经过标准化,不能使用。
- 如果有少部分负值,则数据取log2时,表达量没有+1,可以使用。
boxplot发现异常样品怎么办(取值范围差异很大)?
- 抛弃异常样品。
exp = limma::normalizeBetweenArrays(exp),将样品拉平。- 取值范围差异较小时,可以使用,也可以不用,没有要求。
代码
rm(list = ls())
#打破下载时间的限制,改前60秒,改后10w秒
options(timeout = 100000)
options(scipen = 20)#不要以科学计数法表示
#传统下载方式
library(GEOquery)
eSet = getGEO("GSE7305", destdir = '.', getGPL = F)
class(eSet)
# eSet现在是list格式,list的长度取决于使用GPL平台的多少,
# 此处只有一个GPL平台,因此list长度为1。
length(eSet)
eSet = eSet[[1]]
class(eSet)
#(1)提取表达矩阵exp (位于assayData@exprs中)
exp <- exprs(eSet)
dim(exp) # 行:基因,列:样品
range(exp) # 看数据范围决定是否需要log,是否有负值,异常值
exp = log2(exp+1) # 需要log才log
boxplot(exp,las = 2) # 看是否有异常样本 (las使x轴label竖过来)
# 如果图中显示,数值范围没有超过8,则可能log2运行了两次,需重新检查。
# 如果图中显示,某个样本的取值范围与其他样品不同,则可能是异常样本。
#(2)提取分组信息 (位于phenoData@data中)
pd <- pData(eSet)
#(3)让exp列名与pd的行名** 顺序完全一致 **
p = identical(rownames(pd),colnames(exp));p
if(!p) { # 如果不一致,则取交集
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
}
#(4)提取芯片平台编号,后面要根据它来找探针注释
gpl_number <- eSet@annotation;gpl_number #(对象提取子集用@)
save(pd,exp,gpl_number,file = "step1output.Rdata")
- 原始数据处理:找不到某些GEO数据的表达矩阵肿么办
数据/代码源自生信技能树课程

















 2332
2332

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









