






问一下大家,我想用blastp找到一个11氨基酸小肽的同源序列,我手里有一些物种(人、鸡、鼠…)的蛋白序列。我把这些物种的蛋白序列合并为了一个fasta文件。
我想筛选出bit-score>20的肽段,于是通过
E
=
q
u
e
r
y
_
s
e
q
u
e
n
c
e
_
l
e
n
g
t
h
∗
t
o
t
a
l
_
d
a
t
a
b
a
s
e
_
l
e
n
g
t
h
/
2
b
i
t
−
s
c
o
r
e
E=query\_sequence\_length*total\_database\_length/2^{bit-score}
E=query_sequence_length∗total_database_length/2bit−score算出了bit-score为20时对应的e_value。
我先对merge.fa进行了搜索,使用了下面的命令:
blastp -db merge.fa -query test.fa -out test.merge.0 -task blastp-short -outfmt 0 -evalue 5283 -word_size 2 -matrix PAM30
又对chicken.fa进行了搜索,使用了下面的命令:
blastp -db chicken.fa -query test.fa -out test.chicken.0 -task blastp-short -outfmt 0 -evalue 44 -word_size 2 -matrix PAM30
但出现了两个问题。
-
首先,bit-score的阈值并不是期望的"20",而是"18"。
chicken 比对结果,箭头示bit-score阈值为18.0
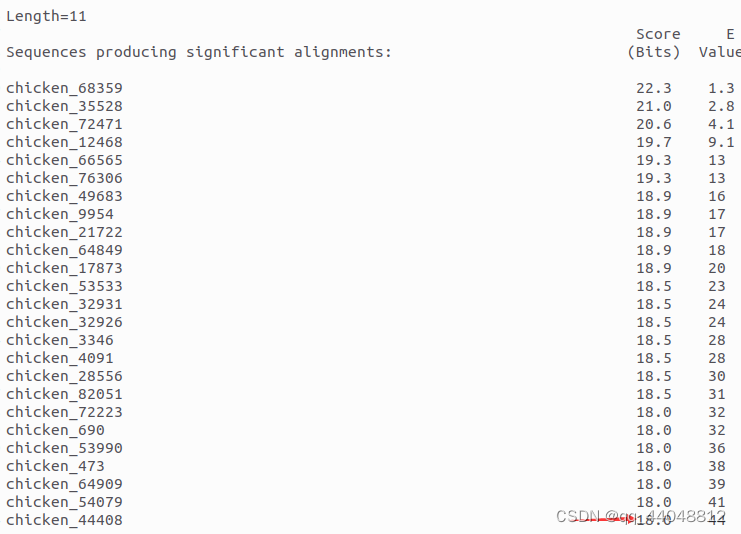
merge 比对结果,箭头示bit-score阈值为18.0

-
其次,我将对chicken的搜索结果与merge的搜索结果进行了比对,发现chicken的搜索结果很多都消失了。
merge中包含chicken的结果




chicken的搜索结果,方框示merge中出现的4个序列,箭头示未出现的多条序列

那些中间的蛋白序列哪去了??














 3272
3272











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









