









前言
- 我们知道通常一个基因会有多条转录本,有时候从ensemble或phytizome上下载的cds或pep文件并没有经过序列过滤
- 而过滤后的序列即仅包含最长转录本有利于我们进一步分析研究,省去后续分析的很多麻烦,例如统计基因数等等
- 因此,我们有必要对序列进行过滤
原始文件
- 那拟南芥为例,其转录本之间的区分是点号.
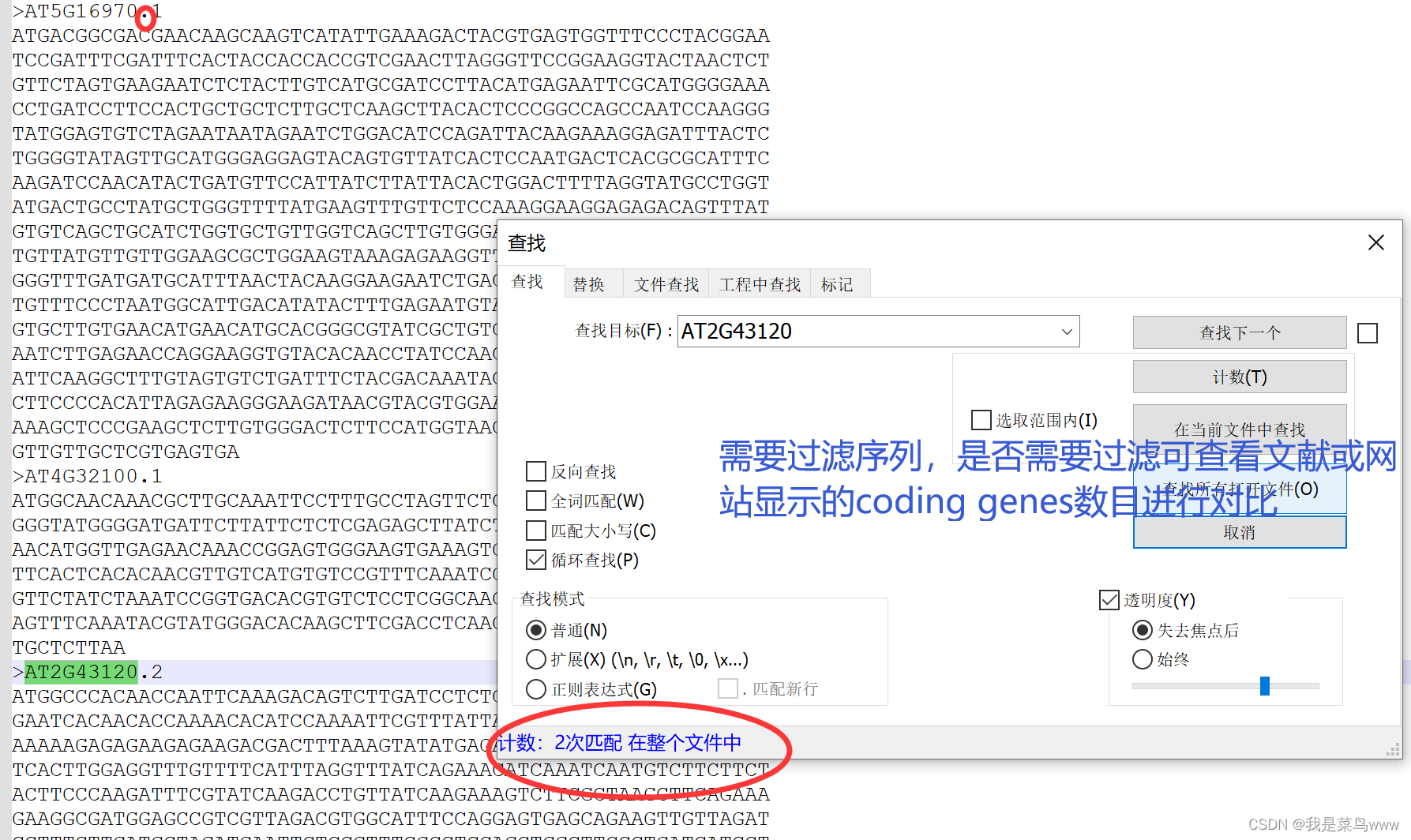
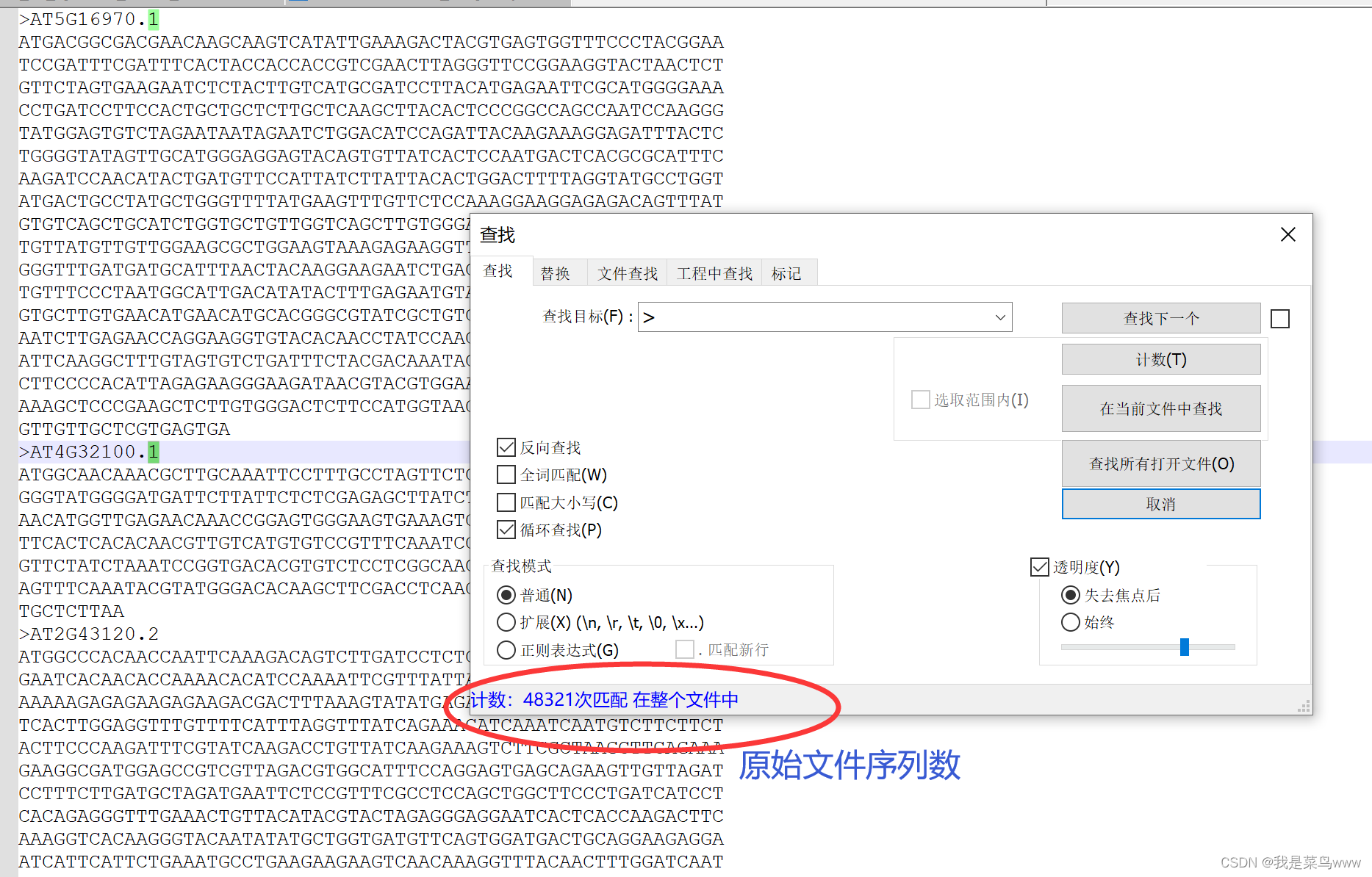
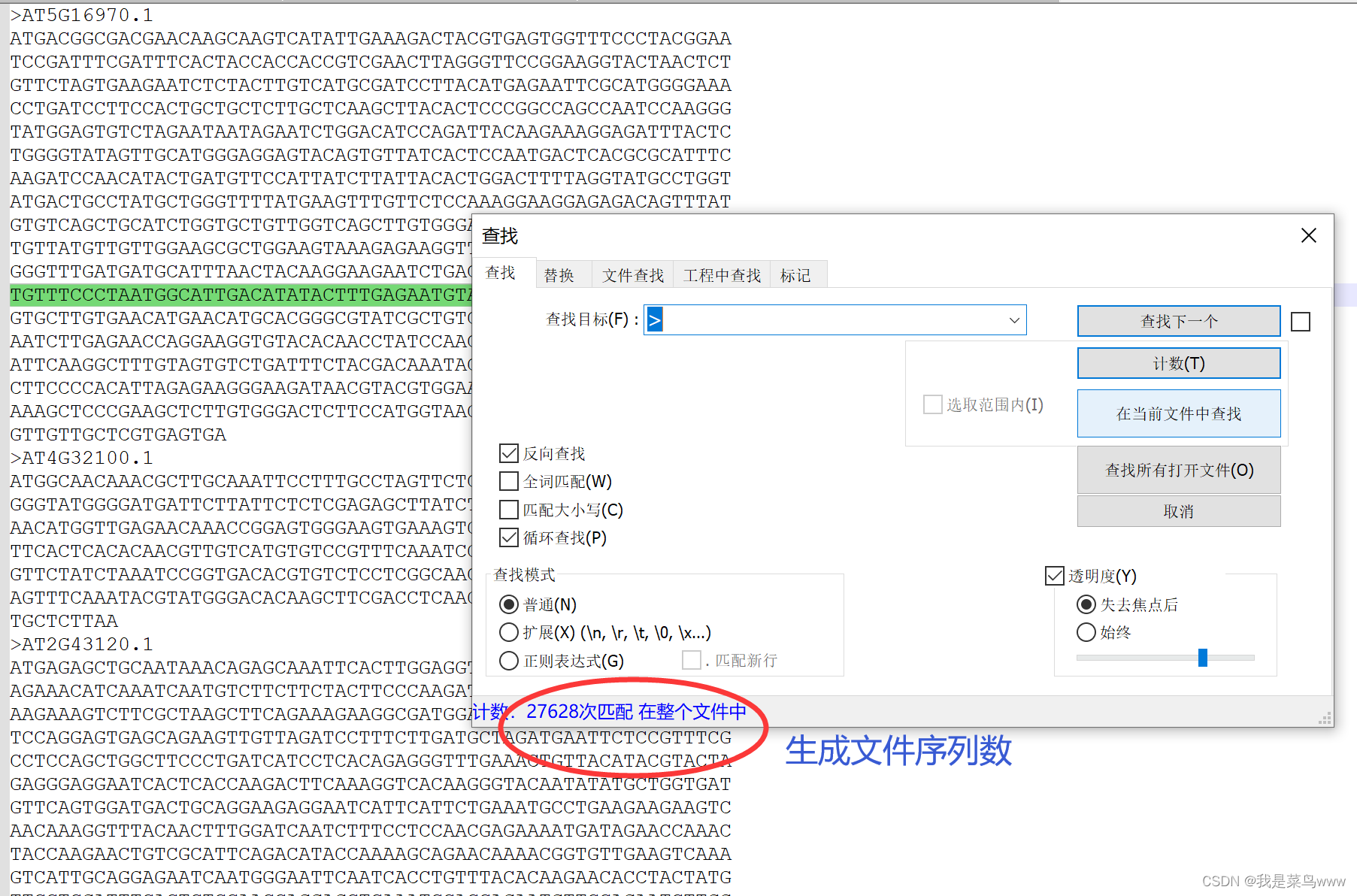
生成文件
脚本实现
- 此脚本适用于批量过滤序列,仅修改脚本中的路径,就可把路径中的fasta文件进行序列过滤
2.此脚本使用于用点号. 区分的序列,如果是其他符号区分如==下划线_==将脚本对应行修改为下划线即可
####仅适用于.区分的转录本
####仅适用于.区分的转录本
from pathlib import Path
p = Path("H:\\徐云峰数据\\CG农科院NP转运体项目\\序列信息\\最长转录本\\selected点")
def select_longest(seq_fa,longest_fa):
#初始化空字典用于存放序列,字典的key是基因名,value存放转录本和序列
#格式为>{gene1:[>gene1.1\nabd\n>gene1.2\neee\nerr\n....]}
seq_dict = {}
with open(seq_fa,'rt') as f1:
for eachline in f1:
if eachline[0] == '>':
gene_name = eachline[1:].split('.')[0]#更改此处.可以选其他符号区分的转录本
if gene_name not in seq_dict.keys():
seq_dict[gene_name] = eachline#此处不加strip(),方便用\n区别转录本和序列
else:
seq_dict[gene_name] += eachline
elif len(eachline.strip()) > 0:
seq_dict[gene_name] += eachline
with open(longest_fa,'wt') as f2:
for i in seq_dict.keys():
transcripts_list = seq_dict[i].split('>')[1:]#此处用切片去除列表的第一个空字符,得到列表 ['gene1\n12\n3456\n', 'gene2\n123\n45\n', 'gene3\n1234\n56789\n']
seq_list= []
for each in transcripts_list:
each = each.split('\n')#单独提取序列
seq = ''.join(each[1:])
seq_list.append(seq)
length = list(map(len,seq_list))
index = length.index(max(length))
f2.write(f'>{transcripts_list[index]}')
def search_fa_longest():
for file in p.iterdir():
if file.suffix == '.fa':
outfile = file.parent / (file.stem + '_longest.fa')
select_longest(file,outfile)
search_fa_longest()
批量文件实现效果如下:
















 6776
6776











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









