







论文地址:TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments
代码地址:https://github.com/lifanchen-simm/transformerCPI
1. Introduction
我们可以将 CPI 问题视为二分类任务,化合物可以被认为是1D序列或分子图(即传统上称为2D结构),蛋白质序列可以被看作1D序列。DeepDTA(Ozturk等人,2018)使用卷积神经网络(CNN)提取化合物和蛋白质的低维实值特征,然后连接两个特征向量并通过完全连接的层来计算最终输出。WideDTA(O¨ztu¨r k et al,2019)和 ConvDTI(Lee et al,2018)遵循了类似的想法,WideDTA 还利用了两个额外的特征,即配体最大共同结构和蛋白质基序和结构域,以提高模型性能。从将化合物结构视为分子图的角度来看,CPI–GNN(Tsubaki等人,2019)和 GraphDTA(Nguyen等人,2019)使用了图神经网络(GNN)和图卷积神经网络(GCN)来代替 CNN 来学习化合物的表示。此外,递归神经网络用于在 DeepAffinity(Karimi等人,2019)中提取化合物和蛋白质的特征向量,Gao等人(2018)和Zheng等人(2020)也将化合物和蛋白质作为序列信息处理。
由于与化学生物学和药物化学的高度相关性,已经开发了许多基于深度学习或机器学习的新模型,在各种数据集上表现出令人满意的性能。然而,很少有人在外部测试或实际应用中评估它们的泛化能力。由于深度学习是一种数据驱动的技术,因此了解模型真正学习的内容并避免意外因素的影响至关重要。最近,谷歌研究人员提出了在机器学习中要避免的三个陷阱(Riley,2019),包括不恰当地拆分数据、隐藏变量和错误的目标。受人工智能行业这些警告的启发,我们想知道基于化学基因组学的CPI建模是否面临着类似的问题,并总结了三个独特的问题。
1.1 使用不合适的数据集
数据是深度学习模型的核心基础,在某种程度上,模型学习的内容主要取决于它所输入的数据集,而不合适的数据集会使模型很容易偏离目标。在基于化学基因组学的CPI建模中,建模的一般目标是基于蛋白质和配体特征的抽象表示预测不同蛋白质和不同化合物之间的相互作用。因此,互作用信息是模型应该从数据集中学习的关键要素。考虑到基于化学基因组学的CPI建模是一项二分类任务,适当设计的数据集应主要由特定配体与蛋白质A相互作用但不与蛋白质B相互作用的实例组成,这迫使模型学习蛋白质信息或相互作用特征,以区分这些实例。
1.2 隐藏配体偏差
DUD-E 和 MUV 数据集中已报告了隐藏的配体偏差问题,这在药物设计领域引起了广泛关注。基于结构的虚拟筛选、基于 3D-CNN 的模型(Chen et al,2019)和基于 DUD-E 数据集训练的其他模型已被指出主要基于配体模式而不是相互作用特征进行预测,导致理论建模和实际应用之间的不匹配。
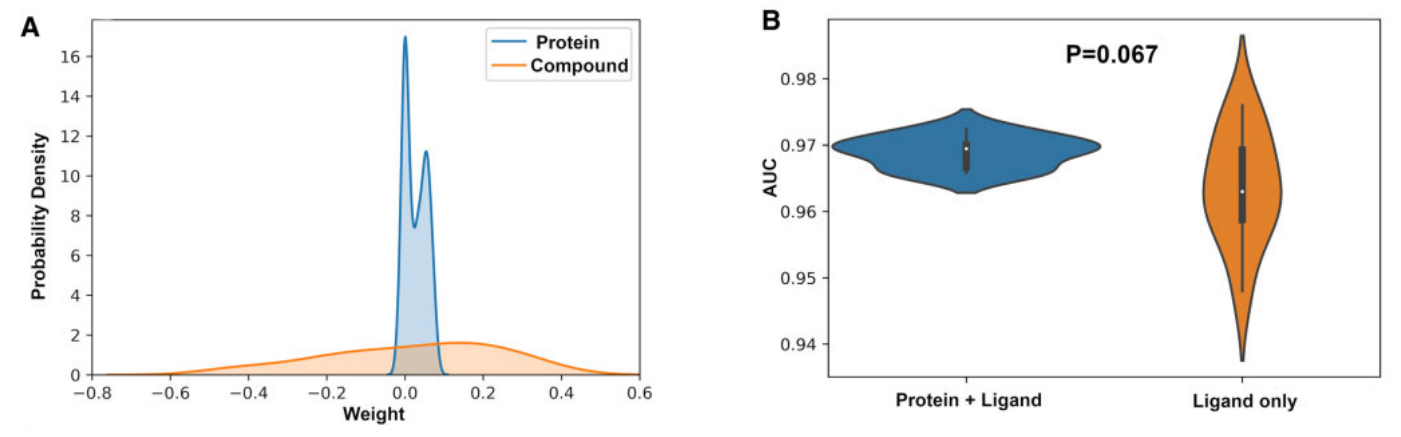
我们想知道基于化学基因组学的 CPI 建模是否面临类似的问题,因此重新访问了以前在人类数据集上训练的典型模型CPI–GNN,作为研究隐藏配体偏差的潜在影响的示例。图1A显示了在人类数据集上训练的 CPI–GNN 模型的权重分布图。用于提取蛋白质特征的 CNN 块的权重显著集中在零,这表明在进行预测时很少考虑蛋白质信息。相反,用于提取复合特征的 GNN 块的权重分布是宽而平坦的。因此,我们认为配体信息与蛋白质信息相比起着压倒性的作用。图1B阐明了仅配体信息的进一步训练及其与原始模型的比较,其中数据集被随机拆分10次,两个模型在10个不同的试验中进行了评估。AUC 分布差异的双样本 t 检验中的 P 值大于0.05,这表明单独使用配体信息可以实现与使用配体和蛋白质信息的原始 CPI–GNN 模型的竞争性能。这些结果强调了配体模式可能误导模型的可能性。
1.3 不恰当地拆分数据集
隐藏配体偏差的风险很难消除,但可以降低。通常,机器学习研究人员将数据随机分成训练集和测试集。然而,使用随机拆分测试集上的常规分类测量,我们不清楚模型是否学习了真实的交互特征或其他意外的隐藏变量,这可能会产生回答错误问题的精确模型。因此,应该根据建模的实际目标及其应用场景来设计测试集。
为了解决这些陷阱,我们提出了一种名为 TransformerCPI 的新型 Transformer 神经网络,构建了专门用于 CPI 建模的新数据集,并引入了更严格的标签反转实验,以评估数据驱动模型是否落入 AI 的常见陷阱。因此,TransformerCPI 在三个公共数据集和两个标签反转数据集上实现了最佳性能。此外,我们进一步研究了 TransformerCPI 的可解释性,通过将注意力权重映射回蛋白质序列和化合物分子来揭示其潜在的预测机制,结果也证实了 TransformerCPI 的自我注意机制在捕获所需的相互作用特征方面是有用的。
2. Materials and methods
2.1 TransformerCPI 的模型架构
我们修改了变压器结构以预测CPI,将化合物和蛋白质视为两种序列。TransformerCPI的概述如图2所示,其中我们保留了 Transformer 的解码器,并修改了其编码器和最终线性层。
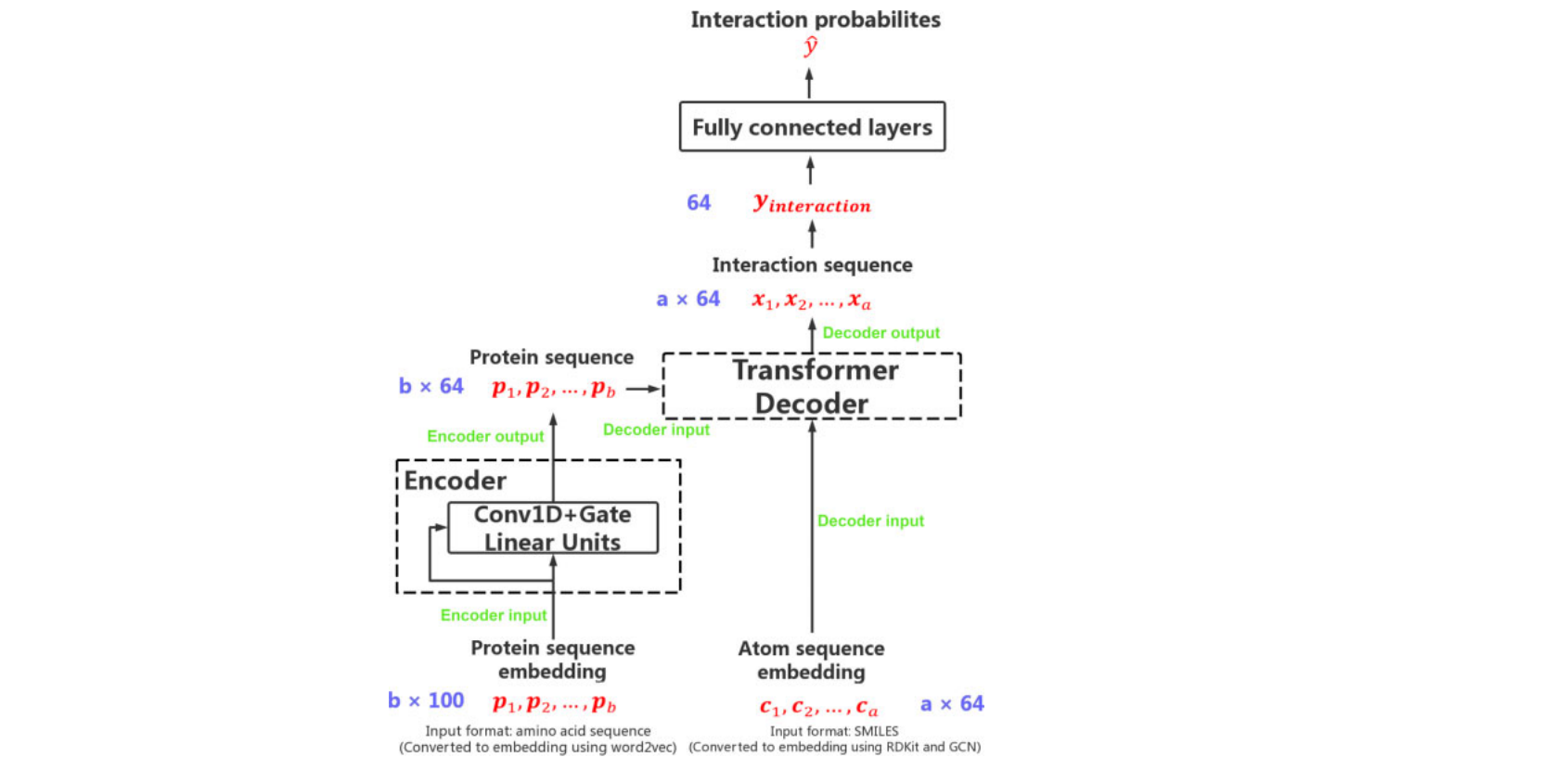
为了将蛋白质序列转换为序列表示,我们首先将蛋白质序列拆分为重叠的 3-gram 氨基酸序列,然后通过预训练方法 word2vec 将所有单词转化为实值嵌入。word2vec 最终可以将单词映射到低维实值向量,其中具有相似语义的单词映射到彼此接近的向量。已经有一些工作应用 word2vec 来表示蛋白质序列,其中恒定长度k(k-mers)的氨基酸序列被拆分为单词,整个氨基酸序列被视为一个文档。我们遵循这些工作来预处理蛋白质序列,并将 UniProt 中的所有人类蛋白质序列作为语料库来预训练 word2vec 模型,并将隐藏维度设置为100。在我们之前构建的大型语料库上训练了30个 epochs 后,可以将蛋白质序列推断为100维实值向量。
然后将蛋白质的序列特征向量传递给编码器来学习蛋白质的更多抽象表示。值得注意的是,我们用相对简单的结构替换了编码器中的原始自我注意层。考虑到传统的 Transformer 架构通常需要大量的训练语料库,并且很容易在小型或中等大小的数据集上过拟合,我们使用了具有 Conv1D 和门控线性单元的门控卷积网络,因为它在我们设计的数据集中表现出更好的性能。门控卷积网络的输入是一系列蛋白质特征向量。我们用等式1来计算隐藏层 h 0 , ⋯ , h L h_0,\cdots,h_L h0,⋯,hL: h 1 ( X ) = ( X ⋅ W 1 + s ) ⊗ σ ( X ⋅ W 2 + t ) (1) h_1(X)=(X·W_1+s)⊗\sigma(X·W_2+t)\tag{1} h1(X)=(X⋅W1+s)⊗σ(X⋅W2+t)(1)其中 X ∈ R n × M 1 X\in R^{n\times M_1} X∈Rn×M1 是隐藏层 h 1 h_1 h1 的输入, W 1 ∈ R k × m 1 × m 2 、 s ∈ R m 2 、 W 2 ∈ R k × m 1 × m 2 、 t ∈ R m 2 W_1\in R^{k\times m_1\times m_2}、s\in R^{m_2}、W_2\in R^{k\times m_1\times m_2}、t\in R^{m_2} W1∈Rk×m1×m2、s∈Rm2、W2∈Rk×m1×m2、t∈Rm2 是待学习的参数, L L L 是隐藏层的数量, n n n 是序列长度, m 1 m_1 m1 和 m 2 m_2 m2 分别是输入特征和隐藏特征的维度, k k k 是 patch_size(卷积核大小), σ \sigma σ 是 sigmoid \text{sigmoid} sigmoid 函数,⊗ 矩阵的对应元素的乘积,门控卷积网络的输出是蛋白质序列的最终特征。在我们的实现中, L = 3 , m 1 = 64 , m 2 = 128 , k = 7 L=3,m_1=64,m_2=128,k=7 L=3,m1=64,m2=128,k=7。Encoder 的输出是蛋白质序列 p 1 , p 2 , ⋯ , p b p_1,p_2,\cdots,p_b p1,p2,⋯,pb, b b b 是蛋白质序列的长度。
每个原子特征最初使用 RDKit python 包表示为大小为34的向量,原子特征列表总结在下表中。然后,我们使用 GCN 通过整合其相邻原子特征来学习每个原子的表示。

GCN 最初是为了解决半监督节点分类问题而设计的,可以将其转换为解决分子表征问题。我们将化合物分子的图表示为
G
=
(
V
,
E
)
G=(V,E)
G=(V,E),其中
V
∈
R
a
×
f
V\in R^{a\times f}
V∈Ra×f 是一个分子中
a
a
a 原子的集合,每个原子表示为
f
f
f 维特征向量,
E
E
E 是分子中共价键的集合,表示为邻接矩阵
A
∈
R
a
×
a
A\in R^{a\times a}
A∈Ra×a。 传播规则如公式2所示:
H
(
l
+
1
)
=
f
(
H
(
l
)
,
A
)
=
σ
(
D
~
−
1
2
A
~
D
~
−
1
2
H
(
l
)
W
3
(
l
)
)
(2)
H^{(l+1)}=f(H^{(l)},A)=\sigma(\widetilde{D}^{-\frac{1}{2}}\widetilde{A}\widetilde{D}^{-\frac{1}{2}}H^{(l)}W_3^{(l)})\tag{2}
H(l+1)=f(H(l),A)=σ(D
−21A
D
−21H(l)W3(l))(2)其中
A
~
=
A
+
I
\widetilde{A}=A+I
A
=A+I,
I
I
I 是单位矩阵,
H
(
l
)
∈
R
a
×
f
H^{(l)}\in R{a\times f}
H(l)∈Ra×f 是第
l
l
l 层的输出,
W
3
(
l
)
∈
R
f
×
f
W_3^{(l)}\in R^{f\times f}
W3(l)∈Rf×f 是第
l
l
l 层的权重矩阵,
D
~
∈
R
a
×
a
\widetilde{D}\in R^{a\times a}
D
∈Ra×a 是
A
~
∈
R
a
×
a
\widetilde{A}\in R{a\times a}
A
∈Ra×a 的对角节点度矩阵。在我们的实现中,
f
=
34
f=34
f=34,GCN 的层数是1。经过 GCN 处理后,得到原子序列
c
1
,
c
2
,
⋯
,
c
a
c_1,c_2,\cdots,c_a
c1,c2,⋯,ca,其中
a
a
a 是原子的数量。
当获得蛋白质序列表示和原子表示时,我们成功地将蛋白质和化合物转换为两个序列,这两个序列符合 Transformer 结构。互作用特征通过 Transformer 的解码器学习。在我们的工作中,蛋白质序列是编码器的输入,而原子序列是解码器的输入,解码器的输出是包含相互作用特征并且与原子序列长度一致的互作用序列。
通过 Decoder 提取特征后,获得了一系列互作用序列
x
1
,
x
2
,
⋯
,
x
a
x_1, x_2, \cdots, x_a
x1,x2,⋯,xa,每个矢量的模数计算公式如下:
x
i
,
=
∣
∣
x
i
∣
∣
2
2
(3)
x_i^,=||x_i||^2_2\tag{3}
xi,=∣∣xi∣∣22(3)其中
i
=
1
,
2
,
⋯
,
a
i=1,2,\cdots,a
i=1,2,⋯,a。每个向量的权重可以用
softmax
\text{softmax}
softmax 函数来计算:
α
i
=
e
x
i
,
∑
i
=
1
a
e
x
i
,
(4)
\alpha_i=\frac{e^{x_i^,}}{\sum_{i=1}^ae^{x_i^,}}\tag{4}
αi=∑i=1aexi,exi,(4)最终的互作用特征用如下公式计算:
y
i
n
t
e
r
a
c
t
i
o
n
=
∑
i
=
1
a
α
i
x
i
(5)
y_{interaction}=\sum_{i=1}^a\alpha_ix_i\tag{5}
yinteraction=i=1∑aαixi(5)最后,将最终的相互作用特征向量
y
i
n
t
e
r
a
c
t
i
o
n
y_{interaction}
yinteraction 送至以下全连接层,并返回化合物与蛋白质相互作用的概率
y
^
\hat{y}
y^。作为传统的二分类任务,我们使用二远交叉熵损失来训练 TransformerCPI 模型:
L
o
s
s
=
−
[
y
log
(
y
^
)
+
(
1
−
y
)
log
(
1
−
y
^
)
]
(6)
Loss=-[y\log(\hat{y})+(1-y)\log(1-\hat{y})]\tag{6}
Loss=−[ylog(y^)+(1−y)log(1−y^)](6)虽然基础 Transformer 模型有六层,隐藏维度为512,但我们将层数从6层减少到3层,隐藏层的维度从512减少到64。蛋白质特征、原子特征、隐藏层和
y
i
n
t
e
r
a
c
t
i
o
n
y_{interaction}
yinteraction 的维度为64。我们保留了最初的八个注意力头,因为这种配置实现了出色的泛化能力。在训练中,我们使用了LookAhead优化器和RAdam优化器,在没有学习速率预热的情况下,解决了Adam优化器引起的最严重的收敛问题。学习率设置为0.0001,批次大小设置为8,梯度在8个批次上累积。

2.2 数据集
2.2.1 公开的数据集
我们在之前的三个基准数据集上比较了我们的模型,即Human dataset、Caenorhabditis elegans dataset 和 BindingDB dataset。Human dataset 和 Caenorhabditis elegans dataset 包括DrugBank 4.1和Matador的正CPI对以及使用系统筛选框架获得的高度可信的负CPI样本。详细而言,Human dataset 包含 1052 种独特化合物和 852 种独特蛋白质之间的 3369 种正相互作用;Caenorhabditis elegans dataset 包含 1434 种独特化合物和 2504 种独特蛋白质之间的 4000 种正相互作用,训练集、有效集和测试集被随机拆分。BindingDB数据集包含来自公共数据库的 39747 个正样本和 31218 个负样本。BindingDB的训练集、有效集和测试集设计良好,测试集包括在训练集中没有配体或蛋白质的CPI对。因此,BindingDB dataset 可以评估模型对未知配体和蛋白质的泛化能力。
2.2.2 标签反转数据集
为了构建专门用于基于化学基因组学的CPI建模的数据集,我们遵循了两条规则:(1)CPI数据来自实验验证的数据集。(2)每个配体都应该存在于这两类中。
许多先前的研究通过CPI对的随机交叉组合或使用基于相似性的方法生成负样本,这可能会引入意外的噪声和未注意到的偏差。
首先,我们从GLASS数据库构建了GPCR数据集。GLASS数据库提供了大量实验验证的GPCR-配体关联,这满足了我们的第一条规则。GLASS数据库使用
I
C
50
IC_{50}
IC50、
K
i
K_i
Ki 和
E
C
50
EC_{50}
EC50作为结合亲和力值,将其转换为负对数
p
I
C
50
pIC_{50}
pIC50、
p
K
i
pK_i
pKi 和
p
E
C
50
pEC_{50}
pEC50。根据早期工作,将阈值设置为6.0来将原始数据集分为正数据集和负数据集。然后,我们选择遵循第二条规则的蛋白质-化合物对来构建最终的GPCR数据集。我们最终的GPCR数据集包括 5359 个配体、356 个蛋白质和其中的 15343 个CPI。
其次,我们基于 KIBA 数据集构建了 Kinase 数据集。KIBA 评分用于结合各种生物活性类型,包括
I
C
50
IC_{50}
IC50、
K
i
K_i
Ki 和
K
d
K_d
Kd,并消除不同生物活性类型之间的不一致性,这大大减少了数据集中的偏差。KIBA 数据集包含从 ChEMBL 和 STITCH 收集的 467 个靶点和 52498 个配体,这确保了KIBA中的数据经过实验验证。鉴于大多数配体只出现一次,我们遵循 SimBoost 过滤原始 KIBA 数据集使其只包含具有至少 10 种相互作用的化合物和蛋白质,共获得 229 种蛋白质和 2111 种化合物。然后,我们使用建议的 KIBA 阈值 12.1 将数据集划分为正集和负集,并选择正集和负集中化合物都存在的蛋白质-化合物对,共产生 1644 种化合物、229 种蛋白质和 111237 种CPI。下表总结了我们构建的 GPCR 数据集和 Kinase 数据集。

为了确认模型实际学习了交互特征并准确评估隐藏变量的影响,我们提出了一个更严格的标签反转实验。标签反转实验的示意图如图所示:
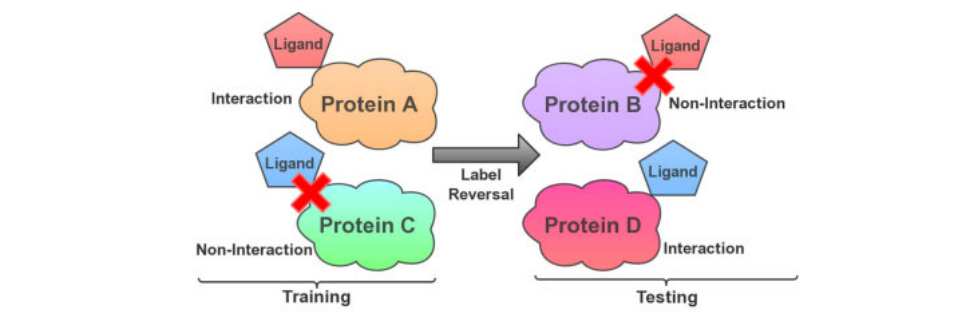
其中训练集中的配体仅出现在一类样品中(正相互作用或负相互作用CPI对),而配体仅在测试集中的相反样品中出现。通过这种方式,该模型被迫利用蛋白质信息来理解相互作用模式,并对那些选择的配体做出相反的预测。如果一个模型只记住配体模式,就不可能做出正确的预测,因为它记住的配体在测试集中有相反的标签。因此,该标签反转实验专门设计用于评估基于化学基因组学的 CPI 模型,并能够指示隐藏配体偏差产生了多大的影响。
对于 GPCR 组和 Kinase 组,我们分别随机选择了 500 和 300 个配体,并将测试集中涉及这些配体的所有负CPI样本合并在一起。此外,我们分别选择了另外 500 个和 300 个配体,并将它们的所有相关正样本汇集在测试集中。在这个实验设计下,我们最终建立了具有1537个相互作用的 GPCR 测试集和具有 19685 个相互作用的 Kinase 测试集。剩余的数据集用于确定超参数,并选择最佳模型来评估标签反转实验。
2.2.3 标签反转数据集的数据分布
在训练模型之前,我们研究了 GPCR 集和 Kinase 集的数据分布。由于每个配体可能出现在多个正和负类中,代表与不同蛋白质相互作用或不相互作用,因此分析了正和负样品中的出现频率。考虑到这个问题,我们计算了每个配体的两类对数比,如下所示,以描述数据分布: log r a t i o ( i ) = log 10 ( N p o s ( i ) N n e g ( i ) ) , i = 1 , 2 , 3 , ⋯ , L (7) \log_{ratio^{(i)}}=\log_{10}\Big(\frac{N_{pos}^{(i)}}{N_{neg}^{(i)}}\Big),i=1,2,3,\cdots,L\tag{7} logratio(i)=log10(Nneg(i)Npos(i)),i=1,2,3,⋯,L(7)其中 KaTeX parse error: Expected '}', got 'EOF' at end of input: {N_{pos}^{(i)} 是第 i i i 个配体的正作用数量,KaTeX parse error: Expected '}', got 'EOF' at end of input: {N_{neg}^{(i)} 是第 i i i 个配体的负作用数量, L L L 是配体的总数。
3. Results and discussion
3.1 公共数据集的性能
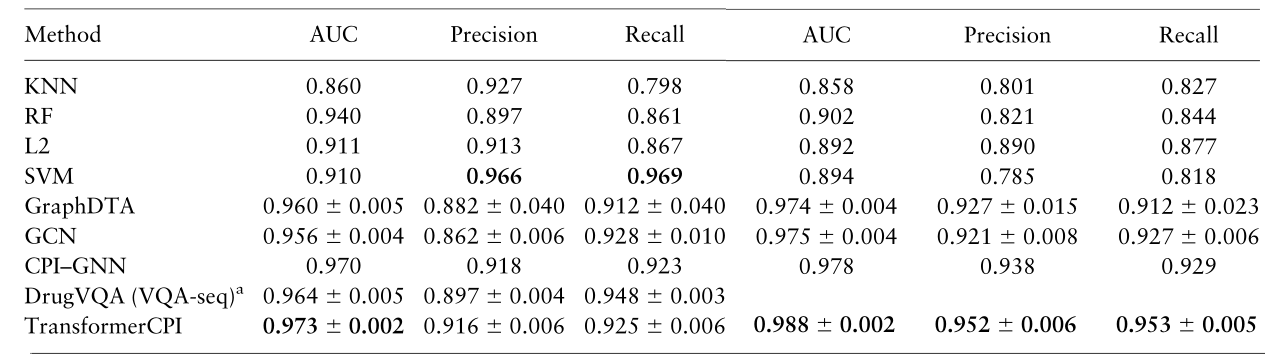
基于序列的模型 CPI–GNN 和 DrugVQA 已经在这些数据集上进行了评估。GraphDTA 最初是为回归任务设计的,在这里,我们将其最后一层改为二分类任务。由于这两个数据集没有这种信息,因此这里不比较依赖于蛋白质的3D结构信息的模型。我们遵循了与 CPI–GNN 相同的训练和评估策略,并使用三种不同的随机种子进行重复,然后使用 DrugVQA 来评估 TransformerCPI,AUC、精度和召回率如下表所示,文献中未提及SVM,这些模型未在BindingDB数据集上进行比较。每个模型的 PRC 和 AUC 如下表所示。TransformerCPI在三个公共数据集上表现优于其他模型。
3.2 标签反转数据集的性能
我们选择 CPI–GNN、GraphDTA 和 GCN 作为参考,并在 AUC 和 PRC 方面比较了 TransformerCPI 与这些模型的性能。为了进行公平的比较,每个模型都在相同的有效集上进行了微调。所有模型在GPCR有效集和Kinase验证集上都实现了相似的性能,然而,在测试集上观察到这些模型之间存在很大的性能差距。尽管这些模型在随机拆分验证集上具有相似的性能,但它们所学到的知识彼此差异很大,这一点通过更严格的标签反转实验得以揭示。在 GPCR 集上,TransformerCPI 在 AUC 和 PRC 方面均优于 CPI–GNN、GraphDTA 和 GCN,显示出捕获化合物和蛋白质之间相互作用特征的能力提高。此外,GraphDTA和GCN在GPCR数据集上取得了良好的性能,这些数据集与 TransformerCPI 接近,但在 Kinase 集上的性能要差得多。相比之下,TransformerCPI 在两个数据集上都取得了最好的性能,显示了它的健壮性和泛化能力。总之,这些结果表明,我们提出的 TransformerCPI 具有学习蛋白质和配体之间相互作用的能力,并且标签反转实验可以有效评估隐藏配体偏差对模型的影响。
3.3 模型的系统依赖性
当比较GPCR集合和Kinase集合之间的结果时,还需要注意的是,TransformerCPI、GraphDTA和GCN在GPCR集合上的表现比Kinase集好得多。我们认为,这种性能差异可能有两个潜在原因。第一个是GPCR集和Kinase集的数据分布不同,导致两个数据集之间的性能差距。第二个原因是,对于TransformerCPI来说,GPCR的序列特征相对更容易学习。对于模型来说,区分相互作用和非相互作用对是一项挑战,因为模型必须学会检测和理解蛋白质序列的微小变化。此外,TransformerCPI的系统依赖性也告诉我们,基于化学基因组学的CPI预测仍有改进的空间,尤其是蛋白质序列的表示。
3.4 消融实验
以前基于化学基因组学的 CPI 模型分别独立地提取配体和蛋白质特征,然后将这两个特征向量连接起来作为输入特征。为了验证 Transformer 中 Encoder-Decoder 架构的作用,我们接下来在相同的标签反转实验中用两个特征向量连接起来代替 Decoder 层并对其评估。显著损害了 TransformerCPI 在 GPCR 集和 Kinase 集上的性能,证明了自注意力机制和编码器-解码器架构在提取两种序列之间的 CPI 特征方面确实起到了关键作用。















 377
377











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









