







摘要
背景:5-甲基胞嘧啶(m5C)是一种关键的转录后修饰,在RNA代谢中起着至关重要的作用。由于生物体中已鉴定的m5C修饰位点大量增加,它们的表观遗传作用变得越来越不为人所知。因此,准确识别m5C修饰位点对于深入了解细胞过程和其他与生物功能相关的机制至关重要。虽然研究人员已经提出了一些传统的计算方法和机器学习算法,但仍然存在一些局限性。在本研究中,我们提出了一种更强大、更可靠的深度学习模型im5C-DSCGA,用于识别人类中新的RNA m5C修饰位点。方法:我们提出的 im5C-DSCGA 模型最初使用三种特征编码方法——one-hot、核苷酸化学性质 (NCP) 和核苷酸密度 (ND)——来提取 RNA 序列中的原始特征并确保剪接;接下来,将原始特征输入到改进的密集连接卷积网络 (DenseNet) 和卷积块注意模块 (CBAM) 机制中以提取高级局部特征;然后,双向门控循环单元 (BGRU) 方法用于从高级局部特征中捕获长期依赖关系并使用 SelfAttention 提取全局特征;最后,使用集成学习并使用全连接对 m5C 位点进行分类和预测。结果:基于深度学习的im5C-DSCGA模型在灵敏度(Sn)、特异度(SP)、准确度(Acc)、马修相关系数(MCC)和曲线下面积(AUC)方面表现良好,在使用三种特征编码方法后,在独立测试数据集中分别产生81.0%、90.8%、85.9%、72.1%和92.6%的值。结论:我们使用五倍交叉验证和独立测试严格评估了im5C-DSCGA的性能,并将其与现有方法进行了比较。在使用独立测试时,MCC指标达到72.1%,比目前最先进的预测方法Deepm5C模型高3.0%。结果表明,im5C-DSCGA模型实现了更准确和稳定的性能,是预测m5C修饰位点的有效工具。据作者所知,这是首次将改进的 DenseNet、BGRU、CBAM Attention 机制和 Self-Attention 机制结合起来预测人类 RNA 中的新型 m5C 位点。
关键词:RNA;5-甲基胞嘧啶位点识别;DenseNet;BGRU;改进的 CBAM 注意力;自注意力;深度学习;集成学习
1. 引言
转录后修饰是生物信息学研究的一个重要领域,目前已鉴定出 170 多种 RNA 修饰 [1]。RNA 会发生多种转录后化学修饰,包括 N1-甲基腺苷 (m1A)、N7 甲基鸟苷 (m7G)、N4-甲基胞嘧啶 (m4C)、5 甲基胞嘧啶 (m5C)、5-羟甲基胞嘧啶 (hm5C) 和 N6-甲基腺苷 (m6A) [2]。其中,5 甲基胞嘧啶 (m5C) 是涉及各种细胞过程的最常见修饰之一。此外,5-甲基胞嘧啶 (m5C) 是一种广泛存在的 mRNA 修饰,发生在 mRNA 转录本的非翻译区 [3,4]。它对许多生物功能至关重要,包括 tRNA 识别、RNA 代谢和应激反应。研究表明,m5C修饰位点在基因表达的多个方面发挥着重要的调控作用,包括核糖体重组、翻译和RNA输出[5]。此外,m5C修饰位点与许多癌症和疾病的发展有关,例如肺癌、肝癌、乳腺癌、常染色体隐性智力低下、肌萎缩侧索硬化症和帕金森病[6– 9]。因此,准确识别RNA中的m5C修饰位点对于揭示相关疾病的表观遗传调控、了解此类修饰的机制和功能具有重要意义。 近年来,RNA修饰受到越来越多的关注,已经开发了许多计算方法来预测RNA中的m5C修饰位点。 一些高通量测序技术,如氧化亚硫酸盐测序 [10]、亚硫酸盐测序 [11]、m5C-RIP-seq [12,13]、Aza-IP-seq 和 miCLIP-seq [14, 15],在过去经常用于识别 RNA 中的 m5C 修饰位点。然而,这些方法测序成本高、耗时长,因此一系列基于机器学习算法的优秀模型被应用于m5C修饰位点,如m5Cpred-SVM[16]、m5Cpred-XS[17]、iRNA5hmC[18]、Staem5[19]等。但机器学习算法只适用于小规模数据集,在数据量较大时可能表现不佳。深度学习算法可以自动处理大规模数据集,能够更好地提取序列的原始特征,从而提升模型的性能。例如,Ali等人[20]开发了iRhm5CNN模型,这是一种高效可靠的用于识别RNA 5hmC位点的计算预测模型,他们使用one-hot编码从RNA序列中提取特征,并利用深度学习中的卷积神经网络结构取得更好的性能。因此,需要寻找或开发一种新颖有效的深度学习方法来识别人类RNA中的m5C修饰位点。 传统的卷积神经网络(CNN)[21,22]在网络深度较深时更容易出现梯度消失问题,而ResNet[23]可以训练更深的CNN模型,达到更高的准确率(Acc)。ResNet在学习过程中参数数量较大,而密集连接卷积网络(DenseNet)[24]通过增强特征传播,大大减少参数数量,缓解梯度消失问题。 这些优势使得使用DenseNet能够以更少的参数和计算成本取得比ResNet更好的性能。例如,Wang等人[25]在2020年设计了一个名为MDCAN-Lys的预测器,利用DenseNet识别赖氨酸乙酰化位点,并在独立的测试数据集上获得了优异的实验结果。随后,Jia 等人 [26] 提出了一种 DeepDN_iGlu 预测器,使用 DenseNet 和注意力机制来预测赖氨酸戊二酰化位点。因此,我们提出的 im5C-DSCGA 模型引入了改进的 DenseNet,以提取 RNA 序列中更高级的局部特征。
传统的循环神经网络(RNN)[27]在学习过程中容易出现梯度消失或梯度爆炸的问题,难以捕捉长RNA序列中各个碱基之间的依赖关系。因此,我们提出的im5C-DSCGA模型引入了双向门控循环单元(BGRU)[28]来捕捉m5C特征之间的长期依赖关系。值得注意的是,深度学习中的注意力机制也经常应用于生物信息学。因此,我们提出的im5C-DSCGA模型引入了改进的卷积块注意力模块(CBAM)注意力[25,29]模块和自注意力[30]模块来捕捉RNA序列中突出的关键特征和全局特征。 2021年,A. EI等人[31]发表了一篇关于m5C修饰位点预测模型的综述。该综述清楚地介绍了目前常用的m5C修饰位点预测模型,并描述了不同模型的评估指标。综上所述,本综述为研究者提供了对m5C修饰位点预测模型的全面认识,为今后的研究和应用提供了宝贵的指导和启示。表1列出了一些m5C修饰位点预测工具的性能,其中NS表示样本数量,WS表示字长。 传统的生物信息学医学实验方法成本高、耗时长,因此开发计算技术并推导出一些优秀的预测器至关重要。我们提出了一种在深度学习背景下识别m5C修饰位点的预测器。 而且,我们的预测器只需要输入一个RNA序列就可以预测这个RNA序列是否是m5C修饰位点,这可以为生物学家提供更便捷的工具,帮助他们更好地理解m5C修饰位点在人类RNA中与基因表达的关系。在本研究中,我们设计了一种基于改进的DenseNet、BGRU、CBAM Attention和Self-Attention组合的混合网络结构,称为im5C-DSCGA模型,用于预测人类RNA中的m5C修饰位点。第 2 节详细介绍了 im5C-DSCGA 模型和每个模块的网络结构。

2. 材料与方法
在这项工作中,我们提出了使用基于深度学习的方法识别人类 RNA 中的 5 甲基胞嘧啶位点。此后,我们将本节中的工作分为四个部分:基准数据集、模型架构、特征提取和评估指标。
2.1 基准数据集
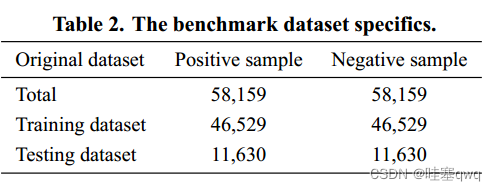
数据集的选取是模型构建的关键部分,本研究采用了Hasan M M 等人[32]的基准数据,以人为研究对象,研究了m5C修饰位点的分布情况,如表2所示。基准数据集具体情况。 为了获得高质量的数据集,他们使用CD-HIT [33]软件去除相似度超过90%的DNA序列。值得注意的是,为了评估模型的稳健性,我们使用与最近研究相同的策略,从原始数据集中随机选择20%(11,630个m5C和11,630个非m5C)并作为独立数据集处理。然而,剩下的 80%(46,559 个 m5C 和 46,559 个非 m5C)被用作训练数据集来开发预测模型。基准数据集的详细信息如表 2 所示。
2.2 模型架构
对于模型架构,我们的讨论将分为两部分。首先,我们将总结 im5C-DSCGA 模型的整体架构,然后详细描述每个模块的结构。
2.2.1 im5C-DSCGA 模型
在本研究中,我们总结了在同一数据集上识别 m5C 修饰位点的预测方法以及 m5C 修饰位点预测的当前进展。虽然 Deepm5C 模型 [32] 取得了相当大的进展,但仍有一些不足之处需要克服。因此,我们设计了一种称为 im5C-DSCGA 的新型深度学习模型来识别人类 RNA 中的 m5C 修饰位点。 图1总结了im5C-DSCGA模型的预测和评估流程设计,该流程由四个部分组成,分别为特征编码、im5C-DSCGA模型框架、集成学习模块和性能评估。在特征编码部分,我们使用了三种编码方式,分别是onehot编码、核苷酸化学性质(NCPÿ
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1606
1606

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









