









Introduction
蛋白质是通过肽键连接在一起的氨基酸链。 由于氨基酸的许多可能组合和链沿着链的多个位置的旋转,这种链的许多构象是可能的。 正是这些构象变化导致蛋白质三维结构的差异。

蛋白质结构预测是生物信息学和理论化学追求的最重要目标之一; 它在医学(例如,药物设计)和生物技术(例如,设计新型酶)中非常重要。[1]
当我们谈论蛋白质的结构时,提到了四种不同的结构水平:初级,次级,三级和四级结构。

蛋白质一级结构是肽或蛋白质中氨基酸的线性序列。
蛋白质二级结构是蛋白质局部区段的三维形式。二级结构元件通常在蛋白质折叠成其三维三级结构之前自发形成中间体。蛋白质和核酸二级结构均可用于辅助多序列比对。
然而,三级结构特别令人感兴趣,因为它描述了蛋白质分子的3D结构,其揭示了非常重要的功能和化学性质,例如蛋白质可以参与哪种化学结合。
仅从其氨基酸序列预测蛋白质三级结构是一个非常具有挑战性的问题,但使用更简单的二级结构定义变得更容易处理。 [2]
我专注于初级和二级结构(SS),更具体地说,使用卷积神经网络(CNN)来预测蛋白质的二级结构,给出它们的主要结构。
Protein Structures and Protein Data
蛋白质的一级结构由其多肽链上的氨基酸序列描述。

有20种天然存在的氨基酸,用一个字母表示法,表示为:'A','C','D','E','F','G','H','I' ,'K','L','M','N','P','Q','R','S','T','V','W','Y'。 'A'代表丙氨酸,'C'代表半胱氨酸,'D'代表天冬氨酸等。第21个字母'X'有时用于表示未知或任何氨基酸。
不是使用一级结构作为存在一种氨基酸的简单指示剂,而是使用了更强大的一级结构表示:蛋白质谱。 这些用于考虑进化邻域并用于模拟蛋白质家族和域。 它们是通过将多个序列比对转换为位置特异性评分系统(PSSM)而构建的。 根据它们在该位置发生的频率对比对中每个位置的氨基酸进行评分。[3]

蛋白质的多肽链通常由约200-300个氨基酸组成,但它可以包含更少或更多。 氨基酸可以在链中的任何位置发生,这意味着即使对于由4个氨基酸组成的链,也有204种可能的不同组合。 在使用的数据集中,平均蛋白质链由208个氨基酸组成。
蛋白质的二级结构决定了蛋白质中局部氨基酸残基的结构状态。 例如,α-螺旋状态形成卷曲的形状,β链形成锯齿状的形状等。蛋白质的二级结构很有意思,因为正如引言中所提到的,它揭示了蛋白质的重要化学性质。 并且因为它可以用于进一步预测它的三级结构。 当预测蛋白质的二级结构时,我们区分3态SS预测和8态SS预测。
对于三态预测,目标是将每种氨基酸分类为:
- α-螺旋,是由'H'表示的常规状态。
- β链,是由'E'表示的常规状态。
- 线圈区域,是由'C'表示的不规则状态。
- 表示上述二级结构的字母不应与表示氨基酸的字母相混淆。
对于8态预测,α-螺旋进一步细分为三种状态:α-螺旋('H'),310螺旋('G')和π-螺旋('I')。 β链被细分为:β链('E')和β新娘('B'),线圈区域被细分为:高曲率环('S'),β转('T' ')和不规则('L')。[2]
E = extended strand, participates in β ladder
B = residue in isolated β-bridge
H = α-helix
G = 3-helix (3-10 helix)
I = 5-helix (π-helix)
T = hydrogen bonded turn
S = bend
_ = loop (any other type)对于该项目的范围,选择了更具挑战性的8状态预测问题。
Dataset
使用的数据集是CullPDB数据集,由6133个蛋白质组成,每个蛋白质具有39900个特征。 6133蛋白质×39900特征可以重塑为6133种蛋白质×700种氨基酸×57种特征。
氨基酸链由700×57矩阵描述,以保持数据大小一致。 700表示肽链,57表示每个氨基酸中的特征数。当到达链的末端时,向量的其余部分将简单地标记为“No Seq”(应用填充)。
在57个特征中,22个代表一级结构(20个氨基酸,1个未知或任何氨基酸,1个'无Seq' - 填充 - ),22个蛋白质谱(与一级结构相同),9个是二级结构(8个)可能的状态,1'没有Seq' - padding-)。
蛋白质谱用于代替氨基酸残基。
有关数据集和下载的更详细说明,请参阅[4]。
在研究的第一阶段,使用整个氨基酸序列作为实例(700×22)来预测整个二级结构(标记)(700×9)。
在第二阶段,沿着序列移动的有限数量元素的局部窗口被用作示例(cnn_width x 21)来预测每个窗口中心的单个位置中的二级结构(8个类别)('在此阶段没有删除Seq'和填充并忽略,因为序列的长度不再相同。
将数据集(6133种蛋白质)随机分为训练(5600),验证(256)和测试(272)组,如[5]所示,结果如下所示。
然而,已经测试了具有不同尺寸的数据集的不同分割,具有相同的结果。
Implementation
该项目是使用带有Tensorflow后端的Keras框架实现的。
已经探索了两种主要方法:
使用整个蛋白质序列(一级结构)作为CNN的实例,具有700×9的输出,即预测的二级结构的序列。
使用有限数量元素的局部窗口作为沿序列移位的CNN的示例,在每个窗口的中心为每个窗口预测单个位置(8个类别)中的二级结构。
1) Whole protein prediction
这个简单的模型由3个主要的1D卷积层组成:
LR = 0.0005
drop_out = 0.3
batch_dim = 64
loss = 'categorical_crossentropy'
# We fix the window size to 11 because the average length of an alpha helix is around eleven residues
# and that of a beta strand is around six.
# See references [6].
m = Sequential()
m.add(Conv1D(128, 11, padding='same', activation='relu', input_shape=(dataset.sequence_len, dataset.amino_acid_residues)))
m.add(Dropout(drop_out))
m.add(Conv1D(64, 11, padding='same', activation='relu'))
m.add(Dropout(drop_out))
m.add(Conv1D(dataset.num_classes, 11, padding='same', activation='softmax'))
opt = optimizers.Adam(lr=LR)
m.compile(optimizer=opt,
loss=loss,
metrics=['accuracy', 'mae'])得到的计算图(来自tensorboard):

这是第一个原型,具有少量参数(125.512可训练参数)。 这种方法的一个主要问题是,填充物增加了较短的序列,仍然影响了损失,计算在整个输出序列上(使用了“tensical_crossentropy”来自tensorwlow的损失)。
这需要创建自定义损失以考虑填充区域的输出,该填充区域对于每个示例具有不同的形状。
很快这种方法被放弃了。
2) Window CNN
This model implementation:
cnn_width = 17
LR = 0.0009 # maybe after some (10-15) epochs reduce it to 0.0008-0.0007
drop_out = 0.38
batch_dim = 64
loss = 'categorical_crossentropy'
m = Sequential()
m.add(Conv1D(128, 5, padding='same', activation='relu', input_shape=(cnn_width, dataset.amino_acid_residues)))
m.add(BatchNormalization())
m.add(Dropout(drop_out))
m.add(Conv1D(128, 3, padding='same', activation='relu'))
m.add(BatchNormalization())
m.add(Dropout(drop_out))
m.add(Conv1D(64, 3, padding='same', activation='relu'))
m.add(BatchNormalization())
m.add(Dropout(drop_out))
m.add(Flatten())
m.add(Dense(128, activation='relu'))
m.add(Dense(32, activation='relu'))
m.add(Dense(dataset.num_classes, activation = 'softmax'))
opt = optimizers.Adam(lr=LR)
m.compile(optimizer=opt,
loss=loss,
metrics=['accuracy', 'mae'])The resulting computation graph (from tensorboard):

窗口的大小选择大于11,因为α螺旋的平均长度大约是11个残基,而β链的平均长度大约是6个(见参考文献[6])。 测试了从11到23的多个均匀尺寸,其中17个产生了最佳结果(性能/训练时间权衡)。
该模型具有232.552个参数(可训练参数:231.912)并且在946494个样本上训练,在120704个样本(窗口)上进行验证。
Results
Window CNN已经过CullPDB数据集拆分训练,如数据集部分所描述的35个时期(在大约6小时内在CPU上)。
学习曲线如下所示:
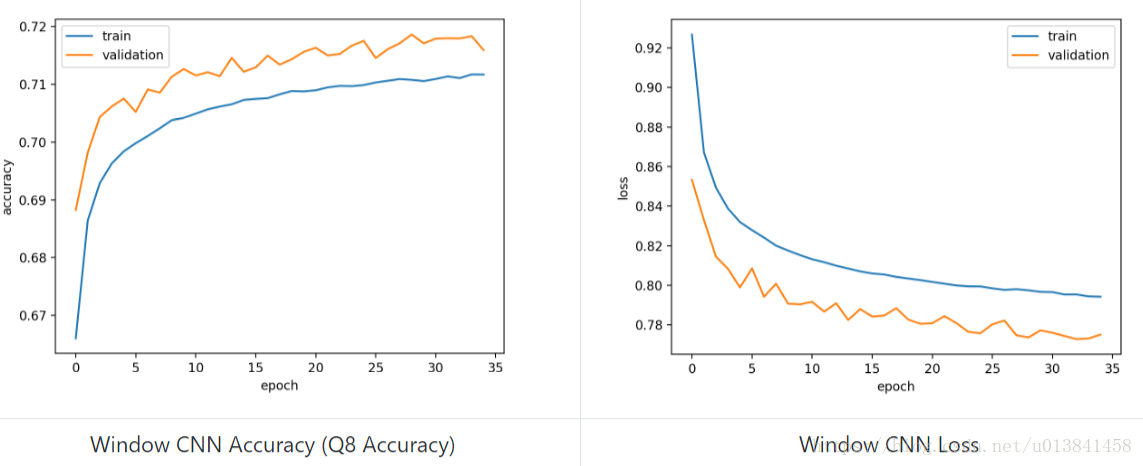
使用该模型获得的测试集的准确度等于0.721522(Q8准确度),这与使用不同技术在[5]和[6]中获得的结果相当。
该模型还使用过滤后的数据集进行了训练:CullPDB6133 +过滤在[4]处可用,并使用公共基准CB513进行测试。 获得的准确度等于0.6833(Q8准确度),再次与[5]和[6]相当。
全蛋白质预测
该模型已经使用CullPDB数据集分割进行了训练,如数据集部分所描述的仅仅20个时期(在大约25分钟内在CPU上)。
学习曲线如下所示(在此模型中,计算损失时不考虑填充,因此得到的值有偏差):
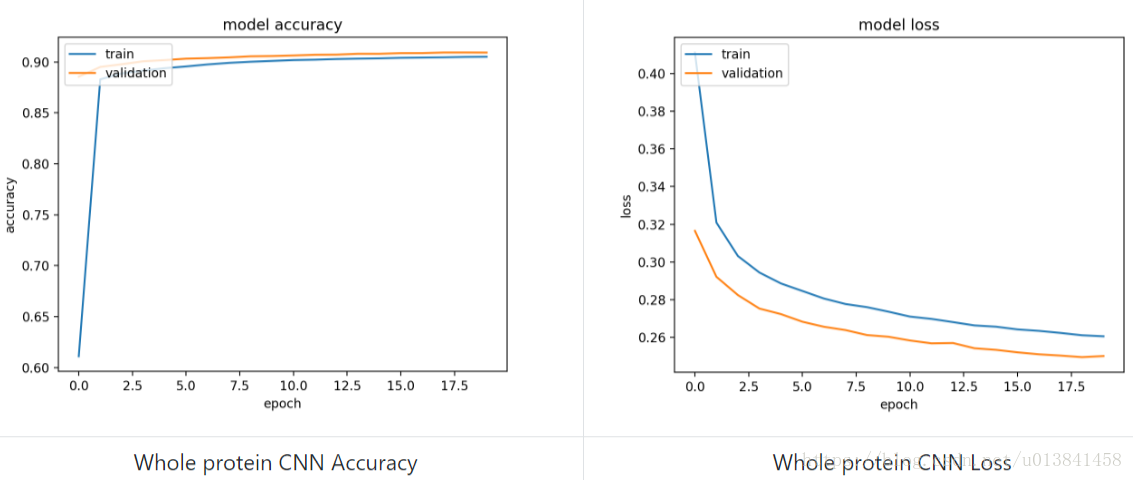
使用该模型实现的测试集的准确度等于0.6966(Q8准确度),这与使用Window CNN在窗口CNN所需的一小部分时间内获得的结果非常接近。
此外,获得过滤数据集训练和CB513数据集测试的准确度等于0.6557(Q8准确度)。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









