





最近(其实是很久之前)因为评论
猫叔:Python如何高效打开超大fasta/fastq/fa/fq文件?zhuanlan.zhihu.com这篇文章,成功被折叠,有感于太多人被各种一时兴起的所谓“纯技术文章”误导,同时一直希望为推动生信领域代码质量提升作出贡献,在此也斗胆写上一篇,希望不会误导大众。
进入NGS时代,高通量测序带来的好处与挑战是并存的,一来我们借此获取了大量的数据,但是,我们就需要面对处理大量数据的挑战,从FASTQ到bam到VCF这样一个最为经典的变异检测流程,对于一个WGS样本来说,我们可能面临超过1TB的数据,如何高效地处理这些数据成为了一个非常大的难题,另外,对于基因组数据,有一个与其他文件不一样的特点,那就是我们常常需要查找特定的基因组位置的数据。经过多年的发展,这些操作其实都已经有了非常高效的最佳实践。私以为对于生信流程来说,我们常常遇到超大型文件的场景有二:
一、遍历整个文件
测序仪的下机数据常常以FASTQ的格式压缩存储,而处理数据的第一步,就是读取整个FASTQ文件以进行测序质量的统计,这要求我们遍历整个FASTQ文件,对于一个测序深度为30X的WGS样本,下机数据量大约100GB,即使今天大多数集群的计算节点是可以有大于100GB的内存的,但是,将整个文件读入内存的做法仍然非常的奢侈且没有任何的意义,何况,100GB是压缩文件,解压后,大约会增长至400GB。依据计算机处理问题的常见思路,我们需要将大问题化解为小问题,通过循环解决小问题来化解大问题。这个问题在读取文件领域的解就是流。流的这个字我认为并没有完全解释流这个理念的全部意味,流的思想是指每次只读取一小部分数据并进行处理,这个处理可以有多个过程,像工厂的流水线一样。以FASTQ文件为例,读取4行(1条reads在文件中占用4行),首先以滑窗法进行trim,传给下一步,统计reads的GC含量,传给下一步……然后再读取下一个4行,再执行相应的操作,不断重复,直至文件结束。这样子的思路就是流的思想,整个过程,我们内存中最大只需要hold住一条reads,而无需读取过多的数据占用内存。
with open('test.fq') as f: # 使用with语句,能够保证文件被正确关闭
while True:
reads_id = f.readline() # 读取第一行,该行是reads的ID,注意不是readlines
if reads_id:
seq = f.readline() # 第二行是序列
comment = f.readline() # 第三行是注释
quality = f.readline() # 第四行是质量值
start, end = trim(quality) # 返回符合质量要求的起始终止位置(假装有这个函数)
count_gc(seq[start:end]) # 统计GC含量(假装有这个函数)
else:
break # 文件结束如上代码所示,就是流概念的经典实现,不需要一次性将整个文件读入内存,而是每次只读取一行,每个循环只读取一个reads,并且处理完后立刻丢弃(Python的垃圾收集自动进行该操作,请自行搜索Python的GC了解更多),无论这个文件有多大,我们的内存只要能hold住一条reads就足够了。当然,Python可以直接对文件对象进行for循环,也是一样的操作,因为文件对象实现了魔术方法__next__ ,使得每次循环返回的就是readline方法的结果。最后,使用with语句,可以创建一个上下文环境,当运行退出with语句所在代码块时,无论是否有异常,我们的文件将被正确关闭(可以通过实现对象的__enter__和__exit__魔术方法,来自定义在进入和退出with代码块时进行的操作,请自行搜索Python魔术方法了解更多)。
其次,由于数据量庞大,正确运用多核性能成为提升数据处理速度的一种重要的解决方案,Python由于GIL锁的存在,导致多线程无法充分发挥,但是并不妨碍我们用多进程进行并行计算(请自行搜索GIL锁并充分区分并行和并发)。此时,流的理念并未失效,甚至可以得到进一步的提升,我们可以借用等待计算进程计算结果的时间进行IO操作,避免硬盘读写瓶颈。文件流在并行计算中的应用可以用经典的生产者消费者模式来描述。
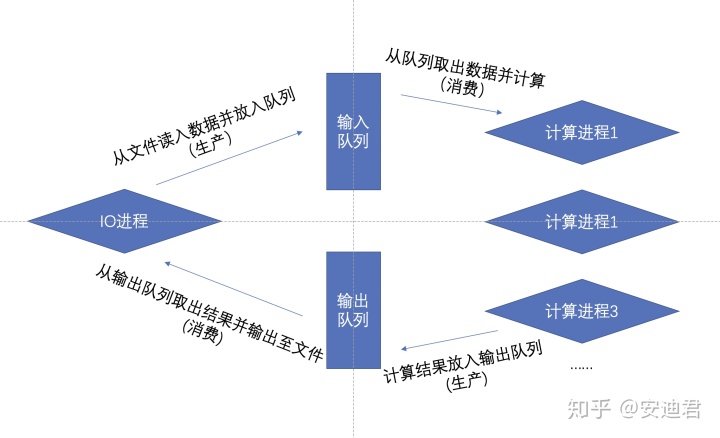
如上图,通过对计算进程与IO进程的分离,IO压力由IO进程承担,而CPU消耗型的多个计算进程同时分别对多个数据进行计算,分散计算压力,通过两个输入输出队列,来实现数据的流转,以加快整个处理过程。这部分代码网上能找到很多小实例,但大多不完整,讲述不清楚,不系统,正好最近改造了illumia开发的spliceAI程序,使其可以多进程并行运行,代码量比较大,就不直接贴出来了,麻烦移步https://github.com/yangmqglobe/SpliceAI/commit/96ef77fef1b01de80680903de5946933965a2902这个commit。与上图的思路一致,通过主进程将变异从文件读入并放入待计算队列,同时几个计算进程从待计算队列取出变异并计算预测结果,结果放入输出队列。主进程在填充输入队列同时也监控输出队列,取出结果并且将结果输出到文件中。通过控制队列的大小来控制总体的内存使用量,一旦输入队列满了,即停止放入,同时不断监控,不让队列为空,使得计算进程无需等待IO阻塞。
二、随机访问基因组位置
生信数据与很多数据的不同在于数据常常是与基因组位置绑定的,而基因组位置与文件位置并不一一绑定。例如,对于bam文件,由于鸟枪法测序本身的随机性,不同位置的深度,即reads的个数不同,我们不能根据位置直接推断给定基因组位置的比对结果位于文件的哪个位置。因而,如果我们想快速跳过我们不想查看的数据,直达我们目标的文件位置,我们就需要有一个索引。索引其实和字典的目录差不多,可以让我们快速知道目标数据所在的位置。根据数据最小单元的大小是否一致,我把索引分为两种:
1、简单等大索引
这种索引的最大特点就是,每个最小的数据单元是等大的,这样我们只需要记录数据不等大的位置点,就可以索引所有的数据。这种索引最经典的例子就是faidx。faidx可以对FASTA文件建立索引,从而使得我们可以快速读取特定基因组位置的序列。其原理是通过记录FASTA文件中所有的contig(或者叫染色体)的起始碱基在文件中的位置(染色体起始前有>和染色体名,为不等大点),由于FASTA文件每行只记录80个碱基,并且每个碱基(最小数据单元)和换行符是等大的(对于utf-8编码的文件,都是1字节),因而,根据基因组位置,我们可以计算目标位置在染色体起始位置之后需要跳过多少个碱基,多少个换行,即我们可以将文件指针直接指向我们需要读取的位置。对于Python来说,其最佳实现是
https://github.com/mdshw5/pyfaidxgithub.com供参考。
2、复杂不等大索引
对于最小数据单元是等大的文件的索引,应该比较好理解,但对于bam和VCF来说,最小数据单元是一条reads或一个变异,它们有不同的质控,不同的标签,因而,它们的每个最小数据单元是不等大的,也就无法使用上面的方法进行索引,此时,就需要介绍另外一个常用的索引软件tabix。tabix软件比较复杂,我们甚至需要从文件的压缩讲起,下面以VCF文件为例,当然,它也可以索引BED、GFF等格式。
常规的VCF文件是文本格式文件,占用存储空间比较大,我们一般需要将其压缩,但是tabix要求不能使用常规的gzip程序进行压缩,而是在按基因组位置排序后使用bgzip压缩。我们可以认为bgzip是gzip的子集,在与gzip压缩格式兼容的情况下,采用BGZF压缩方法,将gzip通常采用更大的block去压缩文件调整为采用不大于64Kb的小block进行压缩(详细信息见http://samtools.sourceforge.net/SAM1.pd,一个block可以认为是文件的一部分,每个block进行压缩和解压需要整个进行,不可再切分),每个block比较小,当我们知道我们寻找的变异就位于某个block的时候,我们不需要解压其他的block,也不需要解压特别大的block,直接解压目标小block就可以了,这样可以大大减少解压带来的消耗(压缩的消耗会增加,但是一般只需要压缩一次,但可能需要解压无数次)。
那么我们如何找到我们想要基因组范围的变异位于哪个block以及它在block中又位于什么位置呢?tabix建立了两个索引,一个称为线性索引(linear index),通过将基因组分为16kb(=2^16b)的等长的窗口,记录每个窗口第一个变异所在的block在文件的位置以及这个变异在整个block中的相对位置。当某个变异落入一个窗口时,我们就可以寻找窗口起始位置所在的block,解压这个block,再从窗口中从前往后筛选我们需要的变异(因为我们已经进行过排序了)。聪明的你应该发现,这种index无法直接准确定位我们需要的数据,和上面的第一种有本质区别。其次,这种index有最差的情况,如果我们的目标变异在窗口的最后,或者有一个长度非常大的结构变异,那我们就需要遍历非常多的变异才能找到我们的目标变异。

因此,tabix建立了第二种索引——分箱索引(binning index),与线性索引不同,分箱索引分为6个等级(上图未全部画出,仅画了4个等级),每个等级的窗口大小不一样,一级只有1个窗口,长512Mb,而人类最长的1号染色体只有249Mb,因而所有的变异都会属于一级的这个唯一的bin,这个bin被编号为0,二级窗口长64Mb,bin编号从1-8,三级窗口长8Mb,编号9-72,以此类推,每次窗口缩短至上一级的八分之一。对于VCF中的每个变异,将其放入其可以完全落入的最小的bin里,例如图中记录A,可以放入编号为1的bin,B放入编号为5的bin,C则放在更小的13号bin。之后,tabix会在索引文件中记录每个bin在VCF所在的block在文件的位置和它在block中的相对位置,根据我们的目标区域,可以推算我们需要在哪些bin里面查找我们的目标变异,遍历这些bin,就可以找到我们的目标变异。此时,会有另外一个问题,那就是我们可能需要遍历非常多的bin,特别是低级别的bin,比如编号0的bin,查询任何区域都需要去查看,这就非常的耗时。
这也是为什么tabix同时生成两种索引的原因,因为tabix程序实际上是同时使用这两种索引的。前面说的,每一个窗口起始位置所在的block在文件的位置和它在block中的相对位置都被记录在索引中,tabix将这两个位置进行了整合,把block所在的文件位置压缩在一个64位的无符号整数较高的48位,把窗口起始位置在block中的相对位置放在了较低的16位,使得这些位置变得可以比较,由于我们的变异已经经过了排序,因而较小的合并位置(tabix的文章把这个位置叫做virtual offset,我觉得不好理解,这里用合并位置代替),就代表较小的基因组位置,较大的合并位置就代表了较大的基因组位置。因此,我们不需要去尝试所有的bin,我们首先用线性索引计算最小的合并位置。再从所有可能的bin中筛选出大于这个最小合并位置的bin。这就使得低等级的bin会被排除,避免做无用功。最近,我为scikit-allel这个Python包实现了使用tabix索引随机访问VCF的代码,使其摆脱了外部调用tabix程序的依赖,具体请移步implement tabix region query natively in python by yangmqglobe · Pull Request #297 · cggh/scikit-allel这个PR,当前这个PR处在草稿阶段,有待更多的测试和调整,仅供参考。
碎碎念
以上就是我个人认为一个bioinfomatics engineer需要知道的处理大型文件的方法。我看过不少生信流程的源代码或者是博客分享,大多数流于表面或者质量参差不齐,虽然本人并非那种秒天秒地的天才,但也希望作出一点贡献,因为我喜欢这个学科,喜欢这个行业。更多的,我希望大家把“Talk is cheap, show me the code”这句口头禅作为论证自己的观点的必要条件,而不是作为反驳别人的观点的论据,共勉。














 803
803











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









