






❝这一篇文章是回答知识星球中一位星友的提问,她的电脑内存有限,无法直接使用所有数据,只能分析部分数据。
❞
数据来源: https://content.cruk.cam.ac.uk/jmlab/atlas_data.tar.gz 解压缩之后,得到下面数据
其中raw_counts.mtx是以「稀疏矩阵」格式存放的表达量数据,文件为6.5G, 用普通的文本编辑器无法打开,我们可以用Linux命令行的less查看数据存放形式
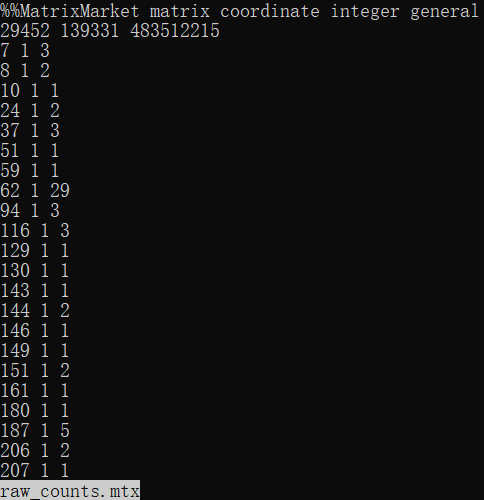
显然这种格式并不是给人类阅读的,它存放的是非零数据的位置及其具体数值。当然,我们也不需要读懂,只需要R语言或者其他编程语言能够加载即可。
R语言的Matrix包的readMM函数就能够读取该文件
mt dim(mt)# [1] 29452 139331# 行为基因,列为细胞这一步时间非常的久,我差不多花了10分钟时间。同时占用内存也非常可观,直接占用了8G左右的内存,不到16G内存的电脑可能根本无法读取。
format(object.size(mt), units = "Mb")# "7377.8 Mb"稀疏矩阵其实和普通矩阵看起来差不多,除了在显示的时候用.来表示0.
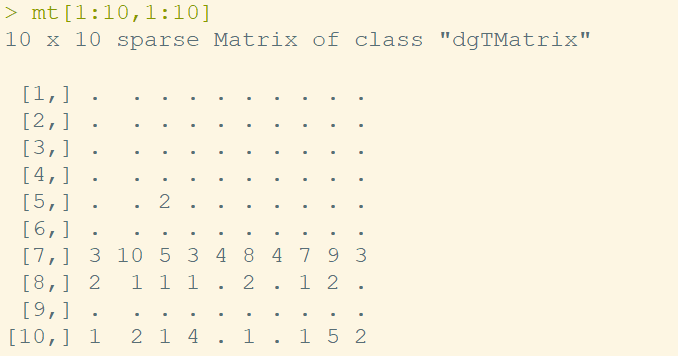
还有一点就是,对于这种量级的数据,我们无法使用R自带的as.data.frame或者as.matrix将其转成普通的数据库或者矩阵,它会直接报错。因此我也不建议对其进行数据转换。
我们发现这里的矩阵并没有行名和列名,这部分信息需要额外从其他文件中读取
bc genes dim(bc)#[1] 139331 1dim(genes)#[1] 29452 2不难发现barcode的行数等于矩阵的列数, gene的行数等于矩阵的行数, 也就是说矩阵的列是细胞,行是基因。
row.names(mt) colnames(mt) 
建议:将此处得到matrix保存为Rds格式,方便后续加载
saveRDS(mt, "raw_matrix.Rds")接下来就是根据元信息来提取对应的细胞,我们以提取"Mesenchyme"细胞为例进行讲解
meta.info sep = "\t", header = TRUE)cell.info cell.info mt.sml format(object.size(mt.sml), units = "Mb")# "280.9 Mb"代码的核心逻辑为提取出对应行的细胞名,然后根据细胞名提取矩阵中的对应列。
过滤后的细胞就可以用作后续分析。不过在开始分析之前,让我们先把原始的矩阵给删掉,因为它实在是太占用内存了。
rm(mt); gc()除了用元信息进行过滤外,你还可以通过随机抽样,从原始数据中抽出部分细胞,这样子也能够在内存吃紧的情况进行后续分析。















 551
551











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









