







Pacific Biosciences单分子实时测序(SMRT)
原始文献——Real-Time DNA Sequencing from Single Polymerase Molecules

Abstract
①We detected the temporal order (时间顺序) of their enzymatic incorporation into a growing DNA strand with zero-mode waveguide nanostructure arrays (零模波导纳米结构阵列), which provide optical observation volume confinement and enable parallel, simultaneous detection of thousands of single-molecule sequencing reactions (成千上万的单分子测序反应能够并行、同步检测).
②Conjugation of fluorophores to the terminal phosphate moiety of the dNTPs allows continuous observation of DNA synthesis over thousands of bases without steric hindrance. ( 将荧光团与dNTPs末端磷酸基结合,可以连续观察上千个碱基的DNA合成,而不受空间位阻。这里可能指的是荧光基团的空间位阻不影响DNA的合成)
③Consensus sequences were generated from the single-molecule reads at 15-fold coverage (15倍的测序深度), showing a median accuracy of 99.3%(准确率中位数是99.3%), with no systematic error beyond fluorophore-dependent error rates (除了与荧光基团相关的错误率外,没有系统误差).
Introduction
①Sanger method: This method relies on the low error rate of DNA polymerases (发挥了DNA聚合酶自身的低错误率), but exploits neither their potential for high catalytic rates (高催化效率) nor high processivity (高持续合成能力).
如何理解:大肠杆菌的DNA 聚合酶I三个功能区,5’→3’ DNA聚合酶活性外,还有5’→3’(去除引物)和3’→5’(检查)的外切核酸酶活性。
Klenow fragment,去除5’→3’外切核酸酶活性,在二代测序合成中应用,效率更高。
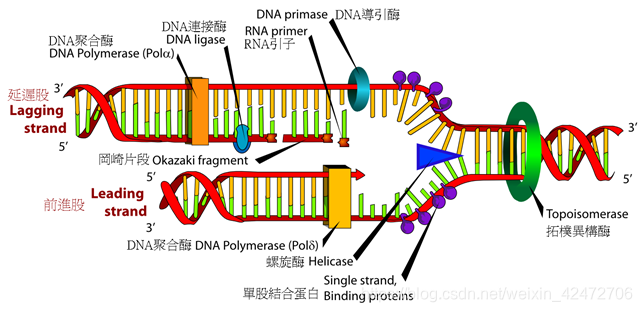
②NGS:However, because these methods all gate enzymatic activity, using various termination approaches, they have not yielded longer sequence reads (limited to ~400 nucleotides), nor do they exploit the high intrinsic rates of polymerase-catalyzed DNA synthesis.
③Goal & Challenge:利用DNA聚合酶作为实时测序的引擎,即使用单个碱基对的分辨率直接观察DNA聚合的过程,这一想法早已提出,但一直难以实现。为了充分利用这些酶的固有速度、保真度和加工性,必须同时应对几个技术挑战。
Ⅰ. 聚合酶合成DNA 的速度随机波动
Ⅱ. dNTP的标记不能影响DNA聚合酶的合成
Ⅲ. 需要保持DNA聚合酶的活性,同时抑制标记的dNTP的非特异性吸附
Ⅳ. 最后,需要一种能够准确检测和区分四种不同标记的dNTPs的仪器。
Methods
Technology
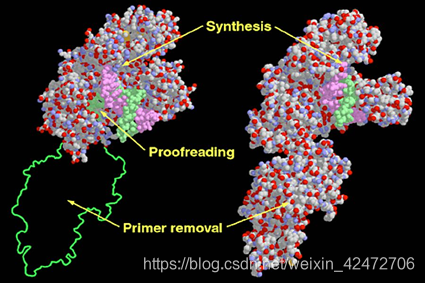
▲when a fluorophore is linked to the terminal phosphate moiety (phospholinked), phosphodiester bond formation catalyzed by the DNA polymerase results in release of the fluorophore from the incorporated nucleotide, thus generating natural, unmodified DNA. (当一个荧光团与末端磷酸基连接时,DNA聚合酶催化的磷酸二酯键形成会使荧光团从合并的核苷酸中释放出来,从而生成天然的、未经修饰的DNA)
▲Φ29 DNA polymerase was selected for these studies because it is a stable, single-subunit enzyme with high speed, accuracy, and processivity (稳定的单亚基酶,具有快速、准确和持续合成能力) that efficiently uses phospholinked dNTPs. It is capable of strand-displacement DNA synthesis and has been used in whole-genome amplification, showing minimal sequencing context bias. (链置换复制模式,就是上次提到的一直循环复制的模式,也被广泛用于全基因组扩增)
▲we reported a surface chemistry that enables selective immobilization of DNA polymerase molecules in the detection zone of ZMW nanostructures with high yield. (使DNA聚合酶分子在ZMW纳米结构检测区域的选择性固定化成为可能)
▲可以测到甲基化,甲基化的碱基脉冲的时常和光谱的特征都会变化,所以可以捕捉。
Structure & Pipeline
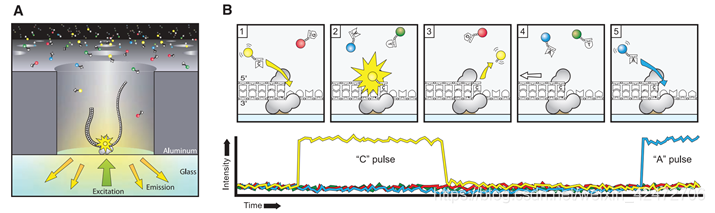
(Fig. A) A single molecule of DNA template-bound Φ29 DNA polymerase is immobilized at the bottom of a ZMW。
(Fig. B) Schematic event sequence of the phospholinked dNTP incorporation cycle, with a corresponding expected time trace of detected fluorescence intensity from the ZMW. (标记的dNTP插入的示意图,以及相对应的从ZMW检测到的荧光强度的预期时间轨迹)
(1) A phospholinked nucleotide forms a cognate association with the template in the polymerase active site, (在聚合酶的活性位点dNTP与模板互补配对)
(2) causing an elevation of the fluorescence output on the corresponding color channel. (催化反应导致相应的)
(3) Phosphodiester bond formation liberates the dye-linker-pyrophosphate product, which diffuses out of the ZMW, thus ending the fluorescence pulse. (磷酸二酯键的形成释放了染料-连接剂-焦磷酸盐产物,该产物从ZMW扩散出去,从而终止了荧光脉冲)
(4) The polymerase translocates to the next position, and (5) the next cognate nucleotide binds the active site beginning the subsequent pulse. ((4)聚合酶转移到下一个位置,(5)下一个同源核苷酸与活性位点结合,开始随后的脉冲。)
PS:A fluorescence pulse is produced by the polymerase retaining the cognate nucleotide with its colorcoded fluorophore in the detection region of the ZMW. It lasts for a period governed principally by the rate of catalysis, and ends upon cleavage of the dye-linker-pyrophosphate group, which quickly diffuses from the ZMW detection region.(荧光脉冲由聚合酶产生,该聚合酶将同源核苷酸及其彩色荧光团保留在ZMW的检测区域。它持续一段主要由催化速率控制的时间,并在染料链接基焦磷酸盐裂解时结束,该裂解迅速从ZMW检测区域扩散。)
The duration of the fluorophore retention is much longer than the time scales associated with diffusion (2 to 10 ms) or noncognate sampling (<1 ms), which manifest as a low and constant background signal. ……The sequence of fluorescence pulses recorded in the plot of intensity versus time is referred to as a read. (荧光信号存在时间长短,区分匹配碱基与游离碱基)
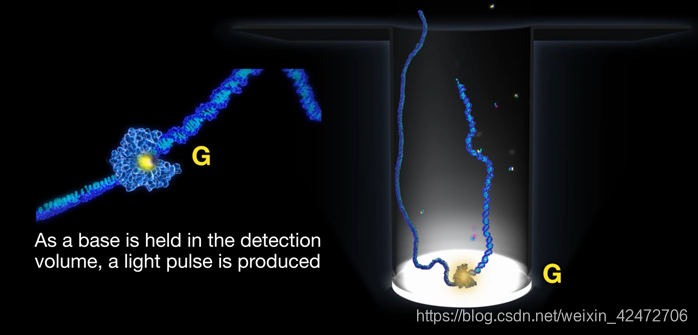
Using synthetic DNA to illustrate approach
To illustrate the principle of our approach to DNA sequencing, we used a synthetic, linear,
single-stranded DNA template with a two-base artificial sequence pattern. (为了说明我们的DNA测序方法的原理,我们使用了一个合成的,线性的,单链DNA模板,但是我们只使用A555-dCTP and A647-dGTP也就是G和C两种dNTP)
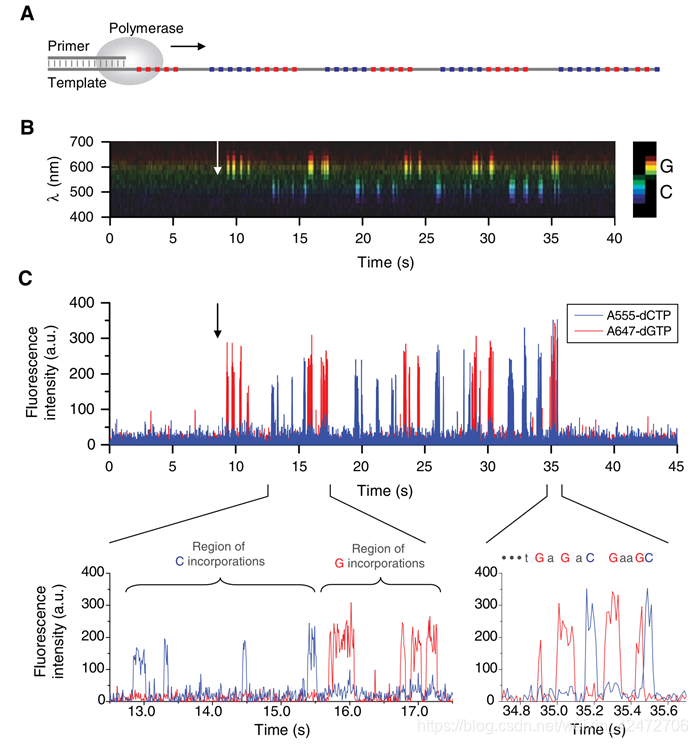
(Fig. A) GC分布的区域
(Fig. B&C)箭头表示引发聚合反应的催化金属离子的加入。
potential of long-read DNA sequencing
we performed a similar two-base signature sequence pattern experiment using a single-stranded 72-base circular DNA template (Fig. 3A). The template was designed such that cytosines were present on only half of the circle, and guanines on the other half. Φ29 DNA polymerase is highly processive (>70,000 bases) without cofactors in bulk reactions. It will carry out multiple laps of DNA strand-displacement synthesis around the circular template. (我们使用单链72碱基圆环状DNA模板进行了双碱基序列模式实验(图3A)。模板的设计使得胞核嘧啶只出现在一半的圆环上,鸟嘌呤则出现在另一半的圆环上。Φ29 DNA聚合酶在没有辅助因子的情况下反应具有高持续性(> 70000个碱基)。它将围绕圆形模板进行多次strand-displacement类型的DNA合成。)
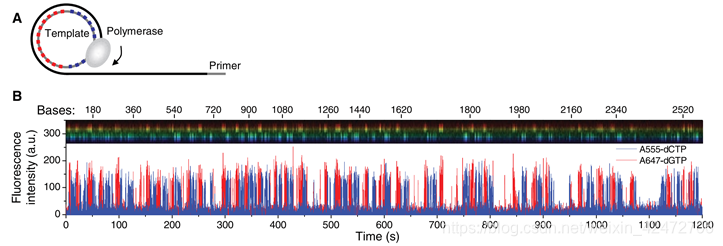
Occasional pauses in DNA polymerization activity are visible as gaps in the trace. The total synthesized DNA length as a function of time (Fig. 3C) shows periods of different persistent polymerization rates during these long reads. Two characteristic polymerization rates of ~2 bases/s and ~4 bases/s were determined, suggesting the existence of different long-lived polymerase modes that occasionally and suddenly interconvert. No spatial correlation in the polymerase speed was observed across a ZMW array. Pulse characteristics underlying these two states were statistically identical, with the exception of a decreased interpulse duration for the faster state (fig. S2). Similar behavior was also observed using different combinations of the fluorophores and bases and for templates with different sequences (fig. S3), which implies that these states are specific neither to the phospholinked dNTPs used nor to the sequence context. (可以看到一些合成中的停顿。总合成DNA长度作为时间的函数(图3C)显示了这些长读过程中不同的持续聚合速率。测定了2个碱基/s和4个碱基/s的两种典型聚合速率,表明存在着不同的持续合成模式,它们之间偶尔会突然发生相互转化。聚合酶速度在ZMW阵列上没有空间相关性。这两种状态下的脉冲特征在统计学上是相同的,除了更快状态下的脉冲间隔时间缩短(图S2)。使用不同的荧光团和碱基组合以及不同序列的模板也观察到类似的行为(图S3),这意味着这些状态既不是特定于所使用的受磷dNTPs,也不是特定于序列上下文)
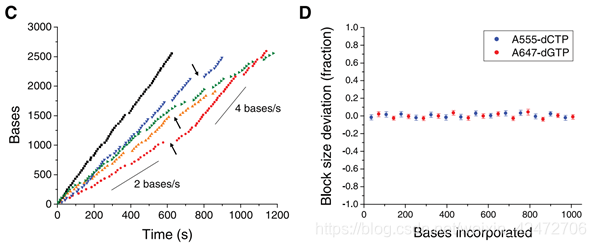
14个循环,1008个碱基,72个一个周期。一个周期上12C个12G。注意只是每个碱基脉冲时间波动很小,而不是每种dNTP的脉冲时间相同)
hence, this sequencing approach maintains accuracy irrespective of read length.
About errors
⚠At optimal loading, the distribution is 36.8% empty ZMWs, 36.8% with just one polymerase, and 26.4% with two or more. (在最佳加载条件下,满足泊松分布,空ZMWs的分布为36.8%,只有一个聚合酶的分布为36.8%,两个或两个以上的分布为26.4%。)
⚠dark nucleotides need not be invoked as a source of error. (暗核苷酸不是错误来源,dNTP足够纯,而且脉冲时间显示应该没有碱基插入事件)
⚠In these data, errors are dominated by deletions, which stem from incorporation events or intervals between them that are too short to be reliably detected. (在这些数据中,错误主要是由删除造成的,删除源于插入事件,它们之间的间隔太短而无法可靠地检测到)图示为A555-dATP脉冲时间与被检测到的概率的比值(黑线),与A555-dATP脉冲时间(蓝线)复合后,得到右图。(从这些数据中,缺失率估计为7.8%,与所观察到的7.4%的核苷酸缺失率一致。)
⚠The majority of insertion errors were caused by dissociation of a cognate nucleotide from the active site before phosphodiester bond formation can occur, resulting in the erroneous duplication of a pulse. (大多数插入错误是由于在磷酸二酯键形成之前,同源核苷酸从活性位点解离,导致错误的重复脉冲)
⚠We have shown that with just 15 molecules, a consensus sequence with 99.3% median accuracy can be formed with no detectable sequence context bias and a uniform error profile within reads. (我们已经证明,只要有15个分子,就可以在不存在可检测序列上下文偏差的情况下形成具有99.3%中位精度的一致序列,并在reads中形成一致的错误表谱)根据上面的实验,不受上下文序列的影响,没有偏好性,只存在随机误差,所以可以通过多次实验避免。

想法:SMRT测序的矛盾点,如果形成Continuous Long Reads (CLR),那么随机误差严重,如果short reads形成Circular Consensus Sequencing (CCS),准确度可以保障,但是又失去了long reads的优势。
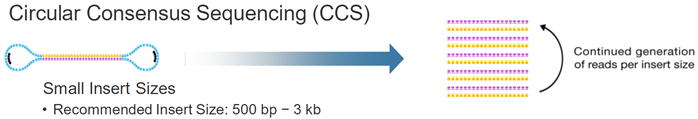
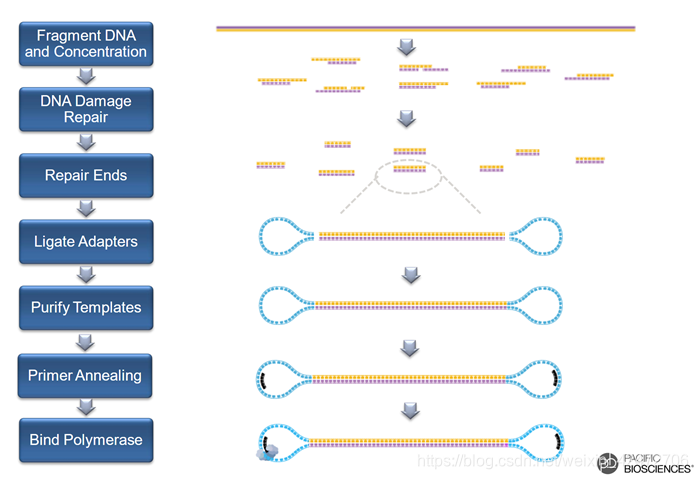
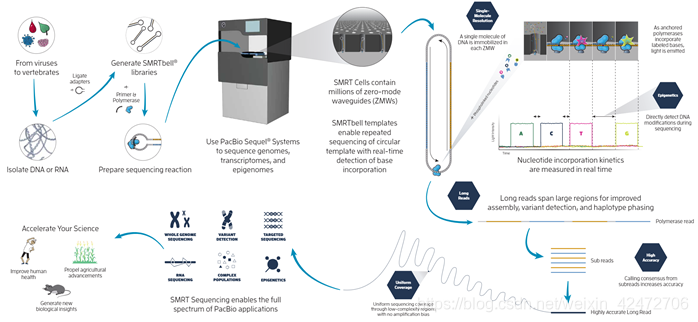
官方SMRT流程文档

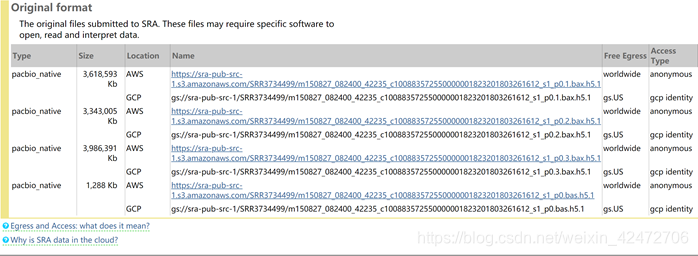
SMRT下机文件示例
DATA sample:DNA N6-adenine methylation in Arabidopsis thaliana
NCBI网址:https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR3734499
bax.h5为后缀的是原始二进制文件,需要Pacbio的软件打开

subreads.fasta / subreads.fastq为后缀的是经一级处理得到的标准格式的碱基文件;
1.m是movie的缩写;2.测序时间,格式为yymmdd_hhmmss;3.代表仪器的编号;4.SMRT Cell Barcode前面提到的接头包含的特殊序列;5和6一般是固定的

read quality (RQ): 对来自一个零模波导孔的subreads的准确度进行预测。有时也用QC Score或Read Score代替。
Pacbio的官方软件内置的算法计算RQ,基于read的脉冲和碱基文件特征(峰值信噪比、平均基QV、脉冲间隔时间等)对读的映射精度进行训练预测。(应该是公司用学习算法预测了一批数据来做预测)
Inset read的筛选包含了序列长短的质量(短序列质量不一定差,但质量差的序列一定短),以及ZMW内p0,p1,p2的状态。
polymerase read: 即高质量测序reads,包含adaptors以及测多次获得multiple subreads。
polymerase reads 是需要进行一定的处理才能获得用于后续分析的。这个过程首先是去除低质量序列和接头序列

第1行跟fasta一致的信息
第2行就是测序得到的序列信息,一般用ATCGN来表示,其中N表示荧光信号干扰无法判断到底是哪个碱基。
第3行以“+”开始,可以储存一些附加信息,一般是空的。
第4行储存的是质量信息,与第2行的碱基序列是一一对应的,其中的每一个符号对应的ASCII值成为phred值,可以简单理解为对应位置碱基的质量值,越大说明测序的质量越好。不同的版本对应的不同。
在测序仪进行测序的时候,会自动根据荧光信号的强弱给出一个参考的测序错误概率(error probability, P)
1.将P取log10之后再乘以-10,得到的结果为Q。
2.把这个Q加上33(前32个不可见字符)或者64(大写字母A是65)转成一个新的数值,称为Phred值,最后把Phred值对应的ASCII字符对应到这个碱基。
e.g. $的ASCII码是36,那么Q=3,错误率P=10^-0.3≈0.5
(的ASCII码是40,那么Q=7,错误率P=10^-0.7≈0.19
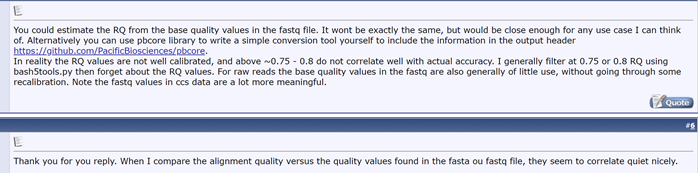
(something interesting on BBS) RQ is close to quality values in fastq file
官方软件

Oxford Nanopore单纳米孔测序

Advantage
- Nanopore DNA sequencing offers the possibility of a label-free, single-molecule approach that can be performed without the need for sample amplification. (纳米孔DNA测序提供了一种无标签、单分子方法的可能性,无需样品扩增即可进行。)
- Like second-generation systems, nanopore technology is amenable to parallelization, and several cost estimates place nanopore sequencing in the $1,000 range for a complete human genome. (一个完整的人类基因组进行纳米孔测序需要花费1000美元。/第二代技术将单倍体人类基因组重新测序到高质量的全部成本(包括仪器、样品制备和人工)目前在10万至100万美元左右。)
- Furthermore, because the sequence quality should be constant throughout a read, long reads from single molecules of DNA will be possible by using nanopores, offering many advantages including the possibility of de novo sequencing, the high-resolution analysis of chromosomal structure variation, and long-range haplotype mapping. (使用纳米孔可以长时间读取单个DNA分子,这提供了许多优势,包括从头测序的可能性、染色体结构变异的高分辨率分析和远程单倍型映射)
Nanopore sequencing principle
Structure
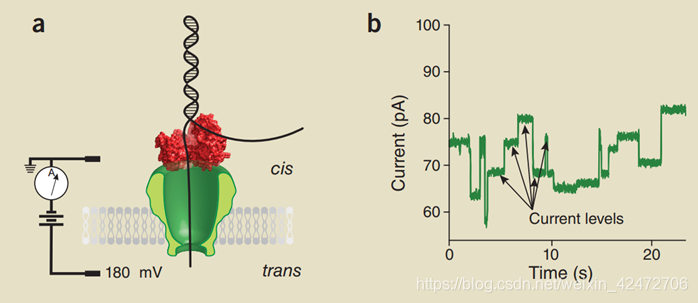
基本的原理就是图中的跨膜的蛋白质(reader)形成一个nanopore,孔径刚好可以穿过一个核苷酸(注意:只是孔径恰好容纳一个碱基,而不是纵向通道容纳一个碱基)。上方的红色蛋白质被称为Motor,本质是解旋酶。膜的电阻率很高几乎不导电,所以电流只能从pore通过,膜两侧是含有离子的溶液,加上电压后,不同的碱基穿过reader导致nanopore不同程度的“堵塞”,从而收集到电流信号。
Pipeline
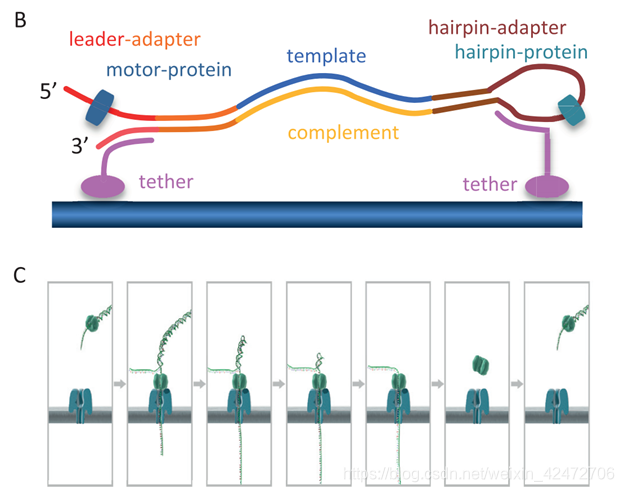
To enable sequencing of both strands, a library is constructed from double-stranded DNA (dsDNA) with a protocol similar to that used for short-read, second-generation platforms. The library preparation chemistries (SQK MAP005 and SQK MAP005.1) used in this study, contain two different adapters that are ligated to the DNA (Figure 1A). The first, the leader adapter , consists of two oligos with partial complementarity that form a Y-shaped structure once annealed. The second, the hairpin adapter , is a single oligo with internal complementarity to form a hairpin structure. Both adapters in the sequencing kit used for this study are preloaded with motor proteins that mediate the movement of DNA through the pore. Another function of the adapters is to guide the DNA fragments to the vicinity of pores via binding to tethering oligos with affinity for the polymer membrane (Figure 1B). Sequencing begins at the single-stranded 5 end of the leader adapter (Figure 1C). (为了实现对两条链的测序,用双链DNA (dsDNA)构建了一个文库。这里用到两个不同的DNA的接头(图1A)。第一个是leader adapter,由两个部分互补的oligos组成,退火后形成y形结构。第二种是hairpin adapter,是一个内部互补的单一寡聚体,形成发卡结构。本研究中使用的测序试剂盒中的两个接头都预先装载了motor蛋白,motor介导DNA通过nanopore。接头的另一个功能链接tether蛋白,引导DNA片段到达孔隙附近(图1B)。测序从leader接头的5’端开始(图1C))
Once the complementary (double-stranded) region of the leader adapter is reached, the motor protein loaded onto the leader adapter unzips the dsDNA, allowing the first strand of the DNA fragment, the template , to be passed into the nanopore one base at a time, while the sensor measures changes in the ionic current. After reaching the hairpin adapter, an additional protein, the hairpin protein , allows the complementary strand of DNA to pass through the nanopore in a similar fashion. (当互补(双链)区域的leader接头到达后,motor蛋白结合到leader接头除,并解旋双链DNA,允许的第一链DNA片段(也就是模板)的碱基依次通过纳米孔,而传感器测量离子电流的变化。到达发夹接头后,另一种蛋白质,即发夹蛋白质,允许互补的DNA链以类似的方式穿过纳米孔)这是一种2D的文库,实际上后来ONT感觉上逐渐淘汰了这种方案。实际上还有1D和1D2两种,区别就是接头不同,1D2可以让第二链紧接着通过,但是存在一定的概率。

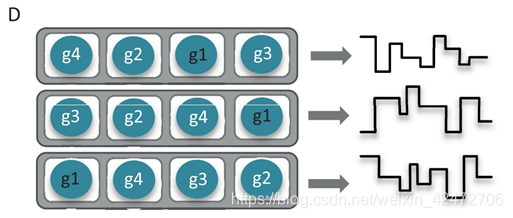
The current MinION flow cell has 512 channels, each connected to 4 wells which may each contain a nanometer-scale biological pore (nanopore) embedded in an electrically-resistant membrane bilayer (Figure 1D). Each channel provides data from one of the four wells at a time, the order of use defined by the allocation of wells to well-groups during an initial mux scan (File S2 Glossary), allowing up to 512 independent DNA molecules to be sequenced simultaneously. (MinION flow cell有512个通道(本质是一个信号放大器),每个通道连接4个孔,每个孔可能包含一个nanopore,但是也可能没有或者两个以上,该生物孔嵌入在耐电双层膜中(图1D)。每个通道同时提供来自四个孔中的一个的数据,会进行预测试,对质量进行排序g1到g4,依次使用8个小时,允许同时对多达512个独立的DNA分子进行排序。)
并不是所有Channel都有信号,ONT公司的数据显示大概只有一半会正常工作,不能工作可能由于pore丢失,或者堵塞;膜有缺口;两个以及以上pore存在,相互干扰。理论上,一个DNA只穿过一次,但是实际上每10min会电流反转一次,激活一些被堵住的小孔,所有可能有些序列被重新推回去,在最新的R9上,pore每秒穿过250个碱基,那么10min大概可以读取150kb,依然是很长的序列了。
Determine the bases & Errors
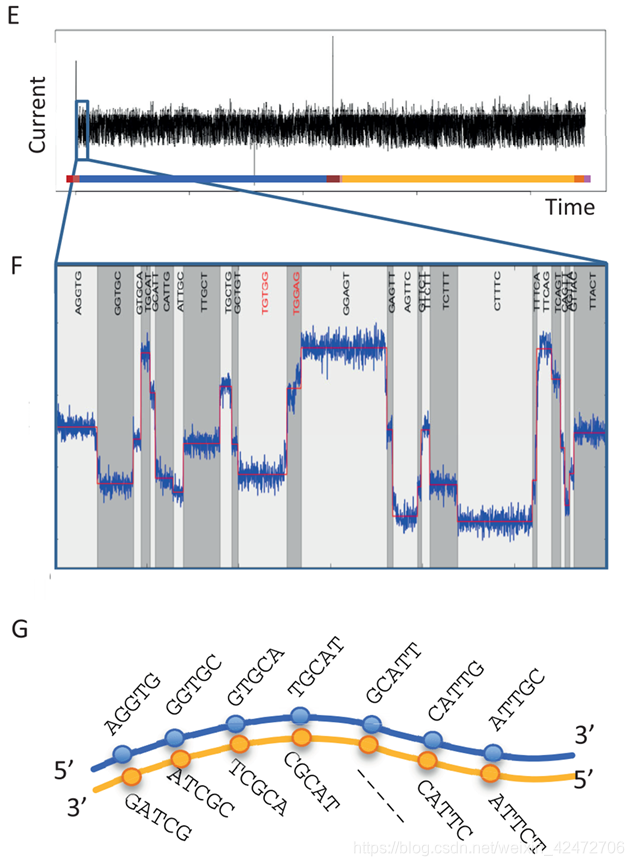
The raw current measurements or the corresponding events, plotted over time, are referred to as a squiggle plot . The base-caller in use at this time modelled the characteristics of 45 (= 1,024) possible 5-mers and base-calling consisted of finding the optimal path (Figure 1G) through a Hidden Markov Model (HMM) of successive 5-mers using a Viterbi algorithm. (原始的电流测量或相应的事件,随着时间的推移,被称为波线图。这个时候使用的base-caller模仿的特点,通过连续隐马尔科夫模型(HMM)训练数据后来推算碱基的排列)对于1D文库准确度85%,1D2可以达到90%.
MinION下机文件fast5
fast5格式存储了nanopore测序过程中全部的输出信息。里面记录着设备运行时全部的信息,包括捕获的电信号值,设备运行时间,电压,温度等等信息。
由于fast5格式可以存储所有的信息,因此,优点就是内容非常全。但是缺点也非常明显,就是占用空间特别大。例如23M左右的碱基序列,存储为fastq格式大概45M,压缩之后大约是23M,而原始的fast5文件则需要占用613M的存储,大约30倍。
一般测序公司只提供fastq格式文件,这个做后续所有的分析其实已经足够了。如果是想要后面重新做碱基校正,可以保留,或者做碱基修饰检验,表观遗传学方面的分析,是需要这个fast5文件的。
个人感觉有点类似于R语言中的S3、S4类,分门别类的储存数据。
Reference
[1] Rank, D., Baybayan, P., Bettman, B., Bibillo, A., Bjornson, K., Chaudhuri, B., … Turner, S. (2009). Real-Time DNA Sequencing from Single Polymerase Molecules. Science, (January), 133–138.
[2] Pacbio官方文档:Pacific Biosciences Glossary of Terms
[3] Pacbio官方文档:Introduction to SMRTbell™ Template Preparation
[4] Pacbio官方文档:Perspective Understanding Accuracy SMRT Sequencing
[5] Pacbio官方文档:Template Preparation
[6] https://zhuanlan.zhihu.com/p/77547922
[7] https://en.wikipedia.org/wiki/FASTQ_format
[8] Pacbio官方文档:SMRT® Analysis Barcoding Overview
[9] Magi, A., Semeraro, R., Mingrino, A., Giusti, B., & D’Aurizio, R. (2017). Nanopore sequencing data analysis: State of the art, applications and challenges. Briefings in Bioinformatics, 19(6), 1256–1272.
[10] Clarke, J., Wu, H. C., Jayasinghe, L., Patel, A., Reid, S., & Bayley, H. (2009). Continuous base identification for single-molecule nanopore DNA sequencing. Nature Nanotechnology, 4(4), 265–270.
[11] Ip CLC, Loose M, Tyson JR et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis[version 1; referees: 2 approved] F1000Research 2015, 4:1075
[12] Ip, C. L. C., Loose, M., Tyson, J. R., de Cesare, M., Brown, B. L., Jain, M., … Olsen, H. E. (2015). MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Research, 4.
[13] https://zhuanlan.zhihu.com/p/91629114
Data
[1] http://datasets.pacb.com/2013/Human10x/READS/index.html
[2] https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2157793
PS
Pacbio Inc (2004年建立,基于康奈尔的研究,以半导体和光子技术结合生物科技,Illumina在18年11月1日收购了Pacbio) & Oxford Nanopore Ltd (牛津大学化学院的Hagan Bayley教授等人建立于2005年)














 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









