






1.fastp对低质量reads过滤和修剪
1.1 conda 下载fastp代码
conda install -c bioconda fastp1.2 fastp使用
确保获得fastq文件,下列为sing-end文件的处理方法。
fastp -i ./SRR3418019/SRR3418019.fastq -o ./SRR3418019/SRR3418019_clean.fastq.gz -z 4 -q 20 -u 30 -n 10 -f 12 -h ./SRR3418019/SRR3418019.fastp.html-
-i: 指定输入文件的路径,这里是./SRR3418019/SRR3418019.fastq,表示fastp将处理这个文件夹下的SRR3418019.fastq文件。 -
-o: 指定输出文件的路径,这里是./SRR3418019/SRR3418019_clean.fastq.gz,表示处理后的读段将被保存为这个路径下的.gz压缩文件。 -
-z: 设置gzip压缩级别,这里是4,表示输出文件将使用中等压缩级别进行压缩,这在速度和压缩比之间提供了一个平衡。 -
-q: 设置平均质量分数阈值,这里是20,表示fastp将移除平均质量分数低于20的读段。 -
-u: 设置不合格碱基百分比阈值,这里是30,表示如果读段中不合格碱基的百分比超过30%,则该读段将被移除。 -
-n: 设置N碱基数量阈值,这里是10,表示如果读段中N碱基的数量超过10个,则该读段将被移除。 -
-f: 设置前端修剪的碱基数量,这里是12,表示fastp将从每条读段的前端移除12个碱基。 -
-h: 指定HTML报告文件的输出路径,这里是./SRR3418019/SRR3418019.fastp.html,表示fastp将生成一个包含详细统计信息的HTML报告文件。
1.2.1 pair-end使用方法
fastp -i reads1.fastq -I reads2.fastq -o reads1_clean.fastq -O reads2_clean.fastq -z 4 -q 20 -u 30 -n 10 - f 15 -t 15 -F15 -T 15 -h **.fastp.html
1.2.2 接头修剪(adapter trimming)
-
默认接头修剪:fastp默认启用adapter trimming,但可以通过
-A或--disable_adapter_trimming参数关闭。 -
自动检测接头:对于单端(SE)数据,fastp通过分析前约100万条读段的尾部来自动检测接头序列。这种检测可能不准确,用户可以通过
-a或--adapter_sequence参数指定接头序列。如果指定了接头序列,将禁用SE数据的自动检测。 -
PE数据的接头检测:对于双端(PE)数据,fastp通过每对读段的重叠分析来检测接头,这种方法既稳健又快速。通常,即使用户知道接头序列,也无需手动输入,因为fastp可以自动处理。但如果需要,用户仍然可以通过
--adapter_sequence和--adapter_sequence_r2参数分别为读段1和读段2指定接头序列。如果fastp无法找到重叠(例如由于低质量碱基),它将使用这些序列分别修剪读段1和读段2的接头。 -
PE数据的自动检测:对于PE数据,默认情况下禁用接头序列自动检测,因为可以通过重叠分析修剪接头。但如果需要,可以通过
--detect_adapter_for_pe(-a)参数启用。
1.2.3 去重复
fastp默认不去重,输入-D可以去重。重复的来源:
-
PCR重复:如果您的RNA-seq样本在文库构建过程中可能经历了PCR扩增,这可能导致某些序列被过度放大,从而产生重复的读段。在这种情况下,去重可以帮助减少这些重复序列对数据分析的影响。
-
生物学重复:在某些情况下,即使没有PCR扩增,也可能存在生物学上的重复,例如在单细胞RNA-seq中。这些重复可能反映了真实的生物学过程,因此在这种情况下,去红可能不是必须的。
-
数据分析流程:在某些数据分析流程中,去重可能会在后续步骤中进行,例如在使用某些转录组组装或定量工具时。在这种情况下,您可能不需要在fastp中进行去重。
-
数据质量:如果您的数据质量非常高,重复序列可能很少,去重可能不是必要的。但如果数据质量较低,或者您观察到了大量的重复序列,那么去红可能是一个有用的步骤。
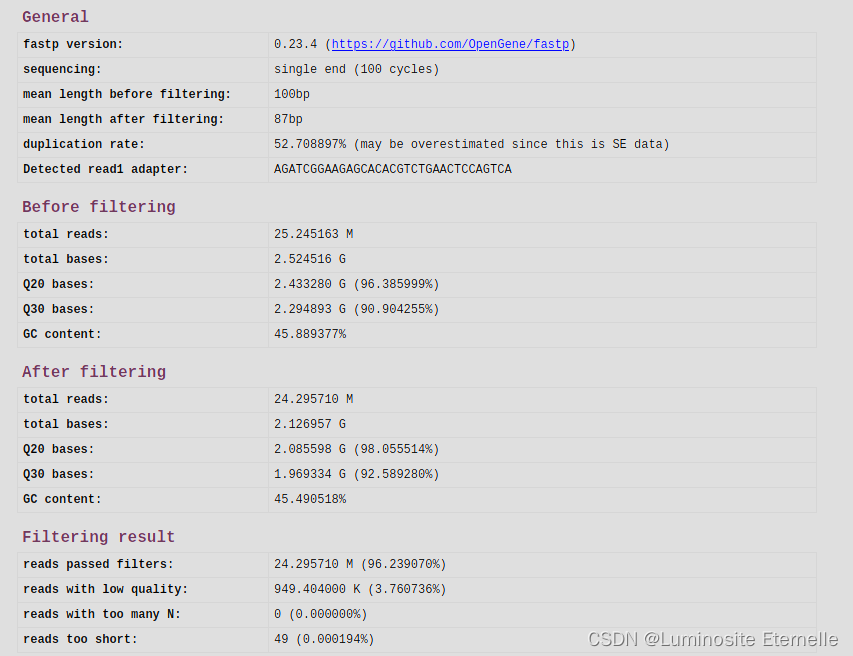
在RNA-seq和宏基因组测序中一般不推荐使用去重复功能,后续在比对过程中会针对重复进行去重。下图左边为未去重(fastp默认不去重),右图去重(-D)。可见去重之后reads被过滤了很多。

2.multiqc
2.1 下载multiqc
conda install -c bioconda multiqc2.2 fastqc对指定fatq文件进行qc
fastqc SRR3418019_clean2.fastq.gz
fastqc SRR3418020_clean2.fastq.gz2.3 multiqc总结报告
multiqc .- .代表在当前文件夹生成报告
--outdir(-o): 在指定的输出目录中创建报告。--title(-i): 报告标题。作为页面页眉打印,如果没有特别指定,则用于文件名。

2.4支持报告类型
-
Assembly:
- SPAdes
- A5 (from A5 pipeline)
- Canu
- Flye
- Miniasm
- Trinity -Velvet
-
Alignment:
- BWA
- Bowtie 1 & 2
- Clustal Omega
- MAFFT
- Muscle
- STAR
-
Variant Calling:
- GATK (haplotype caller)
- Samtools (mpileup)
- VarScan
- FreeBayes
- BCFtools (csq)
-
RNA-Seq:
- HTSeq
- featureCounts
- Salmon
- StringTie
- DESeq2
- edgeR
-
ChIP-Seq:
- MACS2
- ChIP-seq quality control (CSQC)
-
Genome Methylation:
- Bismark
- MethyKit
-
Genome Assembly Evaluation:
- QUAST
- Assemblathon
-
Metagenomics:
- Mothur
- VSEARCH
-
Proteomics:
- OpenMS (FeatureXML)
- MaxQuant
- PeptideProphet
-
Other Tools:
- FastQC
- MultiQC (self-analysis)
- Picard
- bedtools
- bcftools
- RSeQC
- qualimap















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









