






系列文章目录
分析策略
通过综合分析RNA-seq分析流程中不同步骤的工具性能发现不同的分析工具和方法对分析结果的准确度和分析时间影响巨大。HISAT2表现出最快的速度和最准确的拼接比对,但是没有STAR的敏感度高。StringTie在速度和准确度上都优于Cufflinks。长读段方法如IDP和Iso-Seq会识别许多短读段技术没有识别到的多外显子转录本,但是会丢失一些单外显子转录本。通常,在从头组装工具中,Oases表现最佳。不经过比对的工具如Salmon-SMEM和kallisto获得了最好的一致性和最高准确度,因此,如果目标不是发现新的转录本,如Salmon-SMEM和kallisto可以作为准确而快速的解决方案。DESeq2和edgeR与不经过比对的工具联用可以获得高准确度的差异表达分析结果。GATK是一个准确的变异位点检测工具,可以与不同的比对工具联用。当与HISAT2或者STAR比对工具联用时,GIREMI可以不依赖基因组准确预测RNA编辑位点。长读段方法如IDP-fusion可以准确预测RNA融合,而短读段方法如FusionCatcher或者SOAPfuse具有更高的灵敏度。通常情况下,整体最好的分析流程对于特定的数据集特定的研究目的来说可能是次优的。比如,对于比对和转录组构建,HISAT2``-StringTie组合具有更高的准确度和更快的速度。但是对于MCF7-300样品来讲,STAR- StringTie组合具有更高的灵敏度。
STAR下游其他工具也有应用,再single cell RNA-seq也有应用,推荐STAR-HTSeq(featureCounts)
1.比对

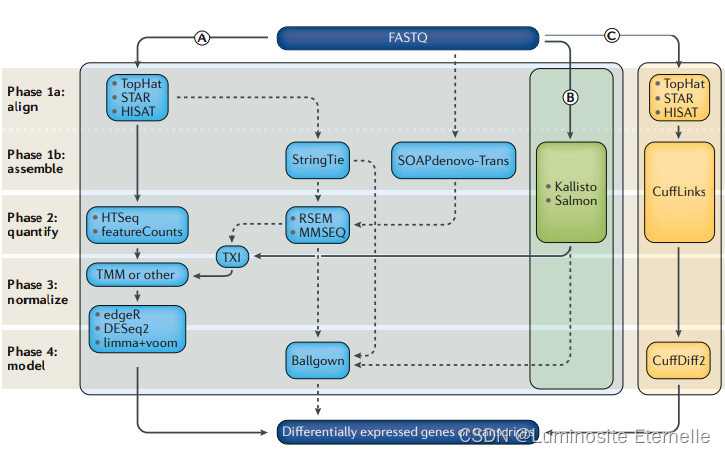
读数映射和转录本鉴定策略。常规 RNA-seq 分析的三种基本策略:
(a) 已有注释的基因组,读数通过间隙映射器映射到基因组。有无注释文件均可进行下一步(新)转录本的发现和定量。
(b) 如果不需要发现新的转录本,则可使用非缺口对齐器将读数映射到参考转录本组。
(c) 如果没有参考基因组,则需要先将读数组装成等位组或转录本。为了进行量化,读数会被映射回新的参考转录本组,然后按照(b)中的方法进行进一步分析,最后按照(a)中的方法对新转录本进行功能注释。每个分析步骤可使用的代表性软件用粗体字标出。
缩写: GFF 通用特征格式,GTF 基因转移格式,RSEM 通过期望最大化进行 RNA 序列分析。
1.1 剪接比对和非剪接比对
剪接比对考虑到了真核生物mRNA前体中的内含子剪接过程。在RNA-Seq实验中,如果测序片段跨越了内含子区域(即包含外显子和内含子序列的片段),则需要将这些片段正确地比对到参考基因组或转录组上。剪接比对工具能够识别并处理这种跨越内含子边界的测序片段,将它们正确地映射到相应的外显子区域。这有助于准确估计基因和转录本的表达水平,并揭示剪接事件对基因表达的影响。
非剪接比对则不考虑内含子剪接事件,它假设测序片段完全来自成熟mRNA的外显子区域。因此,非剪接比对工具会将测序片段直接比对到参考基因组的外显子序列上,而不考虑跨越内含子边界的情况。这种方法在处理原核生物的RNA-Seq数据或某些特定的真核生物RNA-Seq数据时可能更加适用,因为这些生物可能没有内含子或内含子剪接事件较少。
选择剪接比对还是非剪接比对取决于研究的目的和数据的特性。对于真核生物,特别是那些存在复杂剪接事件的生物,剪接比对通常是更合适的选择,因为它能够更准确地反映基因和转录本的表达情况。然而,如果研究关注的是原核生物或剪接事件较少的真核生物,或者数据的特性使得剪接比对变得不切实际,或者只关注已知基因或转录本那么非剪接比对可能是一个更合适的选择。
2.转录组组装
如果没有可用的包含已知外显子边界的高质量基因组注释,或者如果希望将reads与转录本(而不是基因)相关联,则需要在比对后执行转录组组装步骤。诸如StringTie和SOAPdenovo-Trans之类的组装工具使用比对reads的gap来推测外显子边界和可能的剪接位点。
转录本重头组装特别适用于参考基因组注释缺失或不完整的物种,或者对异常转录本感兴趣(例如在肿瘤组织中)的研究。转录组组装方法受益于双端测序和/或更长的reads的使用,增加跨越splice junctions的可能性。但是,通常不需要从RNA-seq数据中从头做转录组组装来确定DGE 。
Sailfish,Kallisto和Salmon,它们将测序reads直接与转录本关联,而无需单独的定量步骤。这些工具在定量高丰度(以及长度更长)的转录本方面表现出很好的性能。但是,它们在定量低丰度或短转录本方面不够准确。
3.定量转录本丰度
reads比对到基因组或转录组后,下一步就是分配给基因或转录本,获得表达矩阵。
3.1 reads计数
转录本定量是RNA-Seq分析中的关键步骤,其中多重比对(即一个reads比对到多个转录本或基因组位置)是一个常见的挑战。处理多重比对reads的策略对于准确估计转录本表达水平至关重要。以下是一些根据多重比对对reads处理方式的转录本定量策略:
- 唯一比对策略:
- 在这种策略中,仅考虑唯一比对到单个转录本或位置的reads。那些比对到多个位置的reads会被丢弃或忽略。如HTSeq-count和Feature count
- 优点:简单直观,易于实现,且结果较为清晰。
- 缺点:可能会损失一些信息,因为丢弃了多重比对的reads。
- 概率分配策略:
- 对于多重比对的reads,根据其在不同转录本或位置上的比对概率进行表达量的分配。如RSEM,cufflinks,stringtie
- 优点:充分利用了所有信息,考虑了reads在不同转录本上的可能性。
- 缺点:计算相对复杂,且可能引入一些不确定性。
- 基于转录本丰度的分配:
- 根据已知或预测的转录本丰度,对多重比对的reads进行分配。
- 优点:考虑了转录本的表达水平,可能更接近真实情况。
- 缺点:需要依赖其他信息或模型来估计转录本丰度。
- eXpress:一个基于转录本丰度的RNA-Seq定量分析工具,它使用最大似然估计方法,并考虑了转录本的已知丰度。
- 使用专门设计的工具:
- 一些转录本定量工具(如RSEM、Cufflinks、Salmon等)已经内置了处理多重比对reads的策略。
- 这些工具通常结合了上述策略中的一种或多种,并提供了额外的优化和特性。
经典的比对分析是将reads比对回参考基因组或者参考转录组,之后估计转录本丰度。如果研究目的是测量已知的和新的转录本丰度,比对回参考基因组后使用Cufflinks和StringTie进行组装和丰度估计。如果使用参考转录组是发现不了新的转录本的,reads可以直接比对到转录组之后使用RSEM和eXpress进行丰度估计。
3.2RNA定量标准化
在RNA定量标准化中,FPKM、RPKM、TMM、DESeq和edgeR等算法和工具各自有其特点和适用场景,选择哪一个取决于你的具体实验设计和分析需求。以下是对这些方法和工具的简要介绍和建议:
- FPKM (Fragments Per Kilobase of transcript per Million mapped reads) 和 RPKM (Reads Per Kilobase of transcript per Million mapped reads):
- 这两者都是基于每百万映射读取数的标准化方法,考虑了基因长度和测序深度对表达量估计的影响。
- 选择FPKM还是RPKM主要取决于你的测序数据类型。如果是单端测序,FPKM和RPKM是一致的;如果是双端测序,则使用FPKM。
- 需要注意的是,虽然RPKM/FPKM被广泛使用,但它们在某些情况下可能不是最佳选择,因为它们没有考虑不同基因或转录本之间的测序效率差异。
- TMM (Trimmed Mean of M-values):
- TMM是RNA-Seq数据分析中常用的一种归一化方法,特别是在edgeR等工具中。
- TMM方法通过计算不同样本间测序深度的比例因子来校正样本间的测序深度差异。
- 当你的数据存在批次效应或其他实验条件差异时,TMM方法可以帮助提高数据的可比性。
- DESeq:
- DESeq是一个基于负二项分布的R包,用于差异表达分析。
- 它不仅提供了数据标准化方法(如使用大小因子进行归一化),还直接进行差异表达测试。
- DESeq特别适用于有生物学重复的实验设计,并能够处理复杂的设计类型,如配对设计或时间序列设计。
- edgeR:
- edgeR是另一个用于差异表达分析的R包,也提供了数据标准化和差异表达测试的功能。
- 它使用TMM方法进行归一化,并采用了基于广义线性模型的统计框架来处理RNA-Seq数据。
- edgeR在处理无生物学重复或低重复的实验数据时表现出色,并且提供了丰富的功能和选项,可以根据具体需求进行灵活调整。















 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









