







关键词:BDE;DFT,Gaussian,量子化学,自由基
键解离能(Bond Dissociation Energy, BDE)是衡量化学反应中键断裂稳定性的重要物理量,指的是将化学键完全断裂所需的能量。在有机化学中,键解离能对于研究分子稳定性、反应机制、自由基反应以及分子间相互作用等方面具有重要意义。因此,精确计算小分子有机化合物的键解离能成为了分子模拟与理论化学研究中的一个重要问题。
键解离能是指在常温常压下,将分子中某个化学键断裂所需要的最小能量。通常,BDE的测定依赖于实验数据,但实验条件的限制、物质的反应性以及实验方法的精确度使得实验获得BDE值存在一定的困难。为了解决这一问题,理论计算方法逐渐成为了研究分子键解离能的有效手段。
对于小分子有机化合物来说,键解离能的研究不仅能帮助了解分子内部的相互作用,还能为设计新型有机材料(如药物分子、聚合物等)、理解催化反应机制以及开发新的反应路线提供理论基础。通过计算键解离能,可以评估分子在不同条件下的稳定性,并预测反应的活性和路径。
通过Gaussian计算的小分子有机化合物的键解离能,对于化学反应的研究具有重要的应用价值。首先,键解离能可以用来评估反应的活化能,预测反应的方向和热力学稳定性。例如,在自由基反应中,自由基的生成通常依赖于键解离能的大小,键解离能较小的键更容易断裂,从而生成自由基。其次,计算得到的键解离能对于分子设计、药物发现和催化剂优化等领域具有指导意义。通过调节分子的结构,可以设计出具有较低键解离能的化合物,从而提高其反应活性或稳定性。
Gaussian计算小分子有机化合物键解离能的难点和挑战
以下是在使用Gaussian研究小分子有机化合物的键解离能(BDE, Bond Dissociation Energy)的一些主要的困难和挑战:
1.计算精度与计算成本的平衡
-
精度要求:键解离能的计算要求高度精确,因为微小的能量差异可能会显著影响反应的预测结果。尤其在涉及到有多个共振结构或者复杂的反应路径时,精确的电子结构计算变得尤为重要。虽然从头算(ab initio)方法和高精度的密度泛函理论(DFT)方法可以提供较高的精度,但这些方法通常计算量非常庞大,尤其是对于大分子系统。
-
计算资源限制:高精度的计算往往伴随着极高的计算成本。尤其是对于含有多个原子和复杂结构的有机化合物,其计算所需的时间和计算资源可能会急剧增加。因此,如何在精度和计算成本之间找到一个平衡点,成为了一个关键问题。
2.溶剂效应的考虑
计算中的溶剂效应,虽然可以通过极性溶剂模型(如PCM,Polarizable Continuum Model)进行模拟,但这种处理也面临以下挑战:
-
溶剂模型的准确性:目前的溶剂模型虽然较为成熟,但对于某些溶剂的极性或溶剂化效应的描述仍然可能存在一定的误差,特别是当溶剂的分子间相互作用较强或者非理想溶剂环境较复杂时。
-
溶剂的多样性:有机分子可能在多种溶剂中表现出不同的反应性,因此,对于不同溶剂环境下的计算,需要采取不同的溶剂模型和方法,而这些模型的选择和调优本身也需要大量的经验和试探。
3.电子结构的复杂性
对于某些有机化合物,特别是那些含有共振结构、过渡态或者自由基中间体的分子,其电子结构非常复杂,计算起来具有较高的挑战性。例如:
-
共振效应:在一些具有共振结构的分子中,键解离的过程可能会受到多个共振形式的影响,因此计算其BDE时,必须考虑到不同共振结构的贡献。
-
自由基生成与稳定性:在键解离过程中,产生的自由基中间体的稳定性会极大地影响反应的能量变化。自由基的生成通常伴随着较大的电子结构变化,这需要通过高精度的计算方法来准确描述其电子态。
4.数据验证与实验对比
尽管理论计算方法得到了广泛应用,但实验数据的验证依然是理论计算结果的最终检验标准。由于实验环境的多样性和复杂性,获得精确的实验BDE值往往非常困难,特别是在极端条件下的反应。因此,计算结果往往需要与已知的实验数据进行对比,以验证模型和计算方法的准确性。
此外,部分小分子有机化合物的BDE可能缺乏直接的实验数据支持,这增加了计算结果的不确定性。
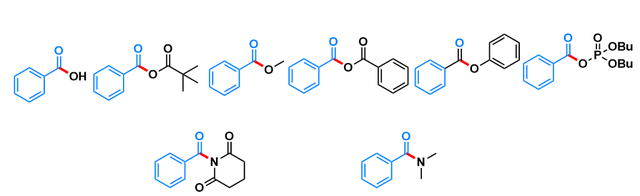
图1几个典型有机化合物
如图1,本案例要算的是这些有机化合物的BDEs,因为这些化合物在偶联反应中是重要的反应底物,其中羧酸,酸酐,酯,磷酸羧酸酸酐,酰胺,扭曲酰胺。这些化合物在偶联反应中分别断的键如图中红色线所示,主要断C-O键和C-N键,计算这些化学键的BDEs对于这些化合物参与的偶联反应的产率和选择性都有非常重要的参考价值。
使用Gaussian键解离能(BDEs)的计算过程:
-
优化分子结构:在进行键解离能的计算之前,需要对分子进行几何优化,找到分子的最低能量构型。这可以通过Gaussian的几何优化功能实现,通常使用DFT方法(wB7XD/def2TZVPP级别)进行优化。优化后的分子结构将为后续的键解离能计算提供基础。
-
计算单键解离能:键解离能的计算需要比较断裂前后两个状态的能量。分子A-B在断裂后生成A和B两个自由基片段,键解离能可以通过以下公式计算:BDE=H(A自由基)+H(B自由基)-H(AB),焓值H=高级别单点能+焓修正量。单点能计算级别:wB7XD/def2TZVPP。特别注意在优化结构和单点能计算时,对于自由基的自旋多重度应该设置为2,因为我们考虑的情况都是化学键的均裂。
-
考虑溶剂效应:在实验中,许多有机化合物的键解离能通常是在溶剂中测量的,因此,计算过程中也需要考虑溶剂效应。采用SMD溶剂模型来模拟溶剂对分子电子结构的影响,从而更加准确地计算溶剂环境下的键解离能。
-
评估计算结果:在完成计算后,通常需要评估计算结果的准确性。通过与实验数据或其他理论方法的结果进行比较,可以验证计算模型的合理性。对于小一点的体系,还可以用更高级别,即精度更高的热力学方法如CBS-QB3,或G4方法计算自由基和分子的热力学数值来计算BDEs。
此次计算的BDEs数值如图2所示:
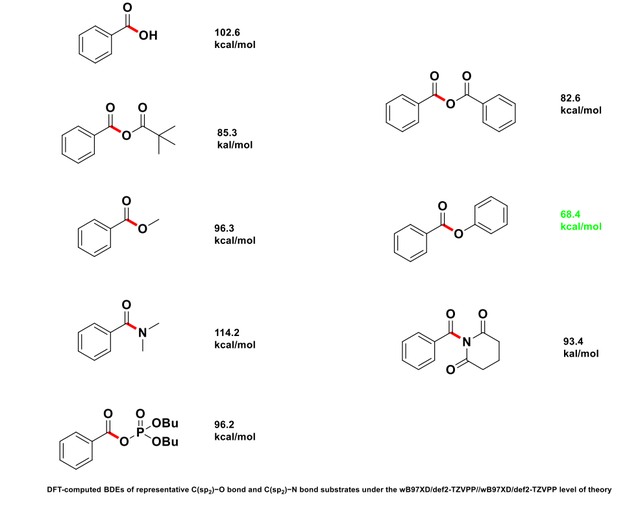
图2 各个化合物的BEDs数值
如图3,这些键解离能(BDEs)差异反映了分子内不同结构因素的影响,特别是共轭效应、自由基的稳定性以及分子间的电子效应。
-
共轭效应:
在苯甲酸(benzoic acid)中,碳-氧键的解离会产生苯甲酰自由基(C6H5CO•),该自由基通过苯环的共轭效应得到一定的稳定性。因此,苯甲酸的BDE较高(102.6 kcal/mol)。对于苯甲酰比瓦烯酸酐(benzoic pivalic anhydride)和苯甲酸酐(benzoic anhydride),尽管它们都含有苯环,氧-碳键解离后生成的自由基同样可能受到电子效应的影响,但它们的BDE较低(82.6 kcal/mol)。这可能是因为在酸酐结构中,氧和碳的电子分布受到更加复杂的电子效应的干扰,导致自由基较苯甲酸中的自由基不太稳定。
-
自由基的稳定性:
在苯甲酸酯(如methyl benzoate、phenyl benzoate)中,产生的自由基(如芳香酰自由基)同样可以通过苯环的π电子进行一定的稳定化,但相较于苯甲酸,酯的碳-氧键解离后的自由基可能稳定性较差,因此BDE稍低。例如,苯基苯甲酸酯(phenyl benzoate)的BDE为68.4 kcal/mol,较低的稳定性可能源于其自由基缺乏像苯甲酸中更强的共轭稳定作用。
-
基团效应:
二丁基磷酸苯甲酰酐(benzoic dibutyl phosphoric anhydride)与苯甲酸酐类似,虽然也含有酰基,生成的自由基受到磷基团的电子效应影响,使得BDE(96.2 kcal/mol)接近苯甲酸,但由于磷的电负性比氧强,可能会对自由基的稳定性产生额外的影响。
-
氮的影响:
在1-苯甲酰哌啶-2,6-二酮(1-benzoylpiperidine-2,6-dione)和N,N-二甲基苯甲酰胺(N,N-dimethylbenzamide)中,碳-氮键的BDE较高(93.4和114.2 kcal/mol)。这些较高的BDE可能与氮的电子效应相关,氮的孤对电子与分子内其他基团的相互作用增强了自由基的稳定性,从而需要更多的能量来断裂这些键。
总结来说,键解离能的差异主要源于共轭效应、自由基稳定性以及基团的电子效应。具有较强共轭效应的分子会具有较高的BDE,而电子效应较强的基团或自由基稳定性差的结构通常表现为较低的BDE。
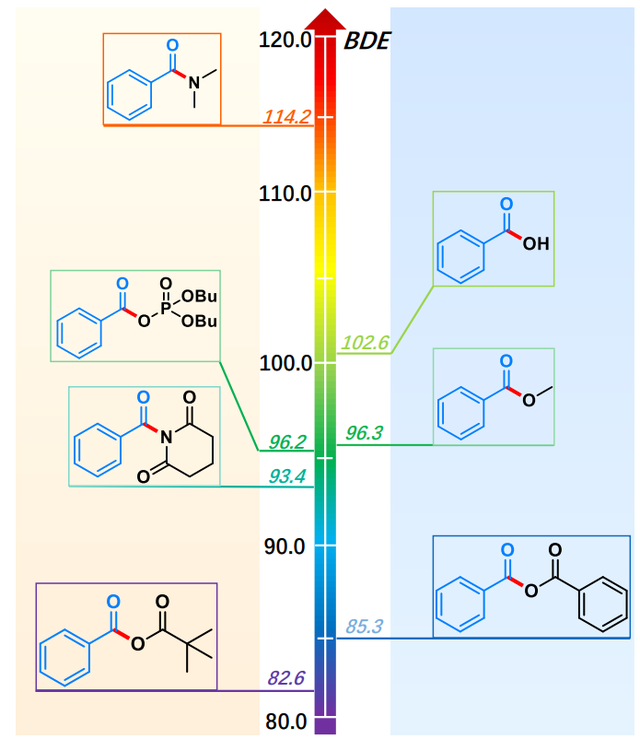
图3 各个化合物的BEDs数值对比图


















 1121
1121

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











