







关键词:pKa,高精度热力学计算,DFT,Gaussian,量子化学
胺类化合物在化学、药物化学和生物化学中扮演着重要角色,它们不仅广泛应用于药物设计、催化反应、环境污染治理等领域,而且其酸碱性质直接影响分子的溶解度、生物利用度和代谢途径。因此,准确预测胺类分子的 pKa 值,对于理解其酸碱行为和调控其化学反应性具有重要意义。pKa 值反映了分子在水溶液中的酸性或碱性强度,通常通过实验测定,但实验方法常常受到溶剂效应、温度、离子强度等因素的影响,且对于复杂分子的测定具有较大的挑战。因此,基于量子化学计算的理论预测成为了近年来研究的热点之一,尤其是高精度计算方法如Gaussian软件的应用,提供了一种可靠的理论工具。
胺类分子通常包含氮原子,并具有一个或多个可接受氢离子的氨基团。胺类分子的酸碱性质通常表现为其在水溶液中与氢离子(H⁺)的结合或解离过程,即酸碱平衡反应。例如,一级胺(RNH₂)在水中与氢离子反应形成铵离子(RNH₃⁺)。在该过程中,胺类分子通过质子化作用(氢离子结合)表现出碱性。胺类分子的 pKa 值定义为其氨基团解离(或接受氢离子)的酸碱平衡常数。
胺类分子的 pKa 值受到多种因素的影响,包括分子的结构(如取代基、环结构、电子效应等)、溶剂的极性、温度和分子的构象变化等。通常,胺类化合物的 pKa 值范围从 9 到 12 左右,但这一范围会因分子内部的电子效应、氢键作用等因素而有所变化。准确预测这些值不仅有助于理解胺类分子的酸碱性,还能帮助设计更具生物活性的分子或优化分子在水相中的稳定性。
Gaussian 软件是目前最广泛使用的量子化学计算工具之一,它能够通过各种量子化学方法模拟分子的结构、性质以及反应机理。在计算 pKa 值时,Gaussian 软件通过密度泛函理论(DFT)、Hartree-Fock (HF) 方法、多体扰动理论(MP2)以及更高精度的配置交互(CCSD)等方法进行分子结构优化和能量计算。这些方法能够提供准确的电子结构信息,为计算 pKa 值提供理论支持。
使用量子化学方法精确计算胺类小分子的 pKa 值时,存在一些技术和理论上的难点。以下是主要的一些挑战:
-
溶剂效应的考虑 胺类分子的 pKa 值是溶液中的酸碱平衡常数,因此溶剂效应对 pKa 值的影响非常大。量子化学方法通常是在气相中进行的,而在气相中的计算无法直接反映溶剂效应。因此,需要引入溶剂化模型(如极性溶剂模型、PCM、SMD 等)来估算溶剂效应,但这些模型往往只能近似溶剂环境,而无法完全精确地模拟真实溶剂对酸碱反应的影响。
-
氢离子的处理 pKa 值的计算本质上是计算氢离子的解离过程。量子化学计算通常要求对氢离子的行为进行精确模拟,尤其是在有机分子中,氢离子的解离常常涉及复杂的相互作用,如氢键、分子间力等。对这些现象的精确建模仍然具有挑战性。
-
构象依赖性 胺类分子的结构可能会影响其酸碱性质。不同的构象可能导致氢离子的解离能量有所不同,因此需要对分子的多种构象进行计算,并找到最低能量构象。然而,在大分子或复杂溶剂环境中,构象的优化和筛选也非常复杂且计算量大。
-
电子相关效应 胺类分子的酸碱行为涉及到电子的重新分布和共振效应。量子化学计算中的电子相关效应(如电子交换-关联能量)对精确预测 pKa 值至关重要,但这些效应需要高精度的波函数方法来处理,如多体效应(多体波函数方法)或高阶的密度泛函方法。高精度的计算通常需要较大的计算资源,特别是在计算分子的多体电子效应时。
-
计算精度和方法的选择 用于量子化学计算的方法(如 DFT、HF、MP2、CCSD 等)各有优缺点。选择合适的计算方法和基组对于得到准确的 pKa 值非常重要。较低级别的方法可能无法捕捉到复杂的电子结构效应,而高精度方法可能会导致计算成本过高,因此需要平衡精度和计算效率。
-
酸碱反应的平衡 计算 pKa 值要求对酸碱平衡进行全面分析,通常涉及到化学反应的自由能变化,这不仅仅是计算单一反应物和产物的能量差异,还要考虑过渡态的能量和解离过程中的所有中间态。在许多情况下,反应路径的选择和过渡态的确定也增加了计算的复杂度。
本案例设计DAP-2H电离的热力学循环,辅助高精度完备基(Complete Basis Set, CBS)方法CBS-QB3计算了DAP-2H分子的酸碱性pKa。计算结果符合实验预期,这得益于我们选用了更昂贵的高精度热力学计算方法。
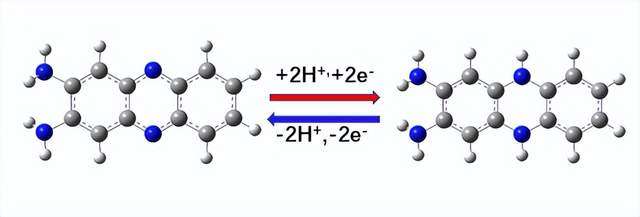
图1 2,3-二氨基吩嗪(DAP) 和DAP2H优化后的结构以及的氧化还原反应
DAP-2H的pKa计算
在如表1所示的计算级别下,分别计算各个分子/离子的吉布斯自由能。根据图2 所述热力学循环,计算DAP-2H的酸碱性pKa。
表1 DAP-2H的pKa计算泛函/基组/溶剂模型
| 分子/离子种类 | 泛函/基组 | 溶剂模型 |
| DAP-2H(gas) | CBS-QB3 | |
| DAP-H-(gas) | CBS-QB3 | |
| DAP-2H(gas) | M052X/6-31G* | |
| DAP-H-(gas) | M052X/6-31G* | |
| DAP-2H(sol) | M052X/6-31G* | SMD(隐式溶剂) |
| DAP-H-(sol) | M052X/6-31G* | SMD(隐式溶剂) |
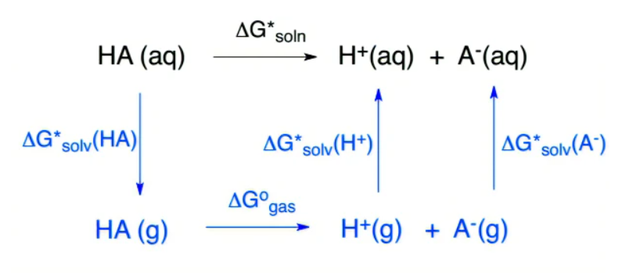
图2 pKa计算的热力学循环
DAP-2H的pKa计算分析
根据图所述热力学循环[54],可得pKa的计算公式如下:
| ΔGsolno=ΔGgaso+ΔGsolv(H+)+ΔGsolv(A-)-ΔGsolv(HA) | (1) |
| pKa=ΔG*soln/RTln(10) | (2) |
| ΔG*soln=ΔGsolno+1.89Δn | (3) |
此计算方法计算pKa时,Ggas(H+)与ΔGsolv(H+)采用了实验值,由下面公式(4)直接计算pKa:
| pKa=[Ggas(A-)-Ggas(HA)+ΔGsolv(A-)-ΔGsolv(HA)-269.0]/1.3644 (4) |
| Ka(5,10-Dihydrophenazine-2,3-diamine) | ||
| Ggas(DAP-2H) | -682.376788 | Haretee |
| Ggas(DAP-H-) | -681.814079 | Haretee |
| Gsolv(DAP-2H) | -0.026523949 | Kcal/mol |
| Gsolv(DAP-H-) | -0.106513784 | Kcal/mol |
| Ggas(H+) | -6.28 | Kcal/mol |
| Gsolv(H+) | -264.61 | Kcal/mol |
| ΔG*soln | 32.62 | Kcal/mol |
| pKa | 23.91 |
表2 DAP-2H的pKa
如表 2 所示,计算得到DAP-2H的pKa为23.91,远大于水的pKa(15.6),证明在水性溶剂环境下,DAP与水分子反应得到DAP-2H分子而稳定存在。


















 1150
1150

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











