




《环球科学》编者按:2024年诺贝尔化学奖颁给了计算蛋白质设计和结构预测领域,《环球科学》2024年10月刊也推出了“AI重塑结构生物学”专题。在诺奖公布前不久,我们独家专访了该专题的审校者、新加坡国立大学计算机科学和生物化学教授、以及癌症科学研究所高级研究员张阳老师,聊了聊蛋白质结构预测领域发展史,以及AlphaFold对该领域的影响。
【注:根据《环球科学》“AI重塑结构生物学”专题的发布时间和编者按,这篇访谈应在2024年诺贝尔化学奖揭晓前的9月或10月完成。虽然该访谈与诺奖公布只是一个时间上的巧合,但是它为本届化学奖提供了一个非常合适的背景注解。特别是,张阳教授在该访谈中对AI以及蛋白质相关的生命科学的一些核心问题进行了高屋建瓴、深入浅出的分析,展现了一个世界顶级结构生物信息学家对这些问题的独到见解,因而显得尤为珍贵。】

华盛顿大学的David Baker教授与AlphaFold的开发者分享2024年诺贝尔化学家奖
黄雨佳(《环球科学》编辑):
张老师好!首先有请张老师简单介绍一下自己的研究背景以及现在的研究方向。
张阳(新加坡国立大学教授):
谢谢。我们的实验室主要致力于人工智能和计算生物学研究。多年来,我们一直专注于蛋白质折叠和结构预测问题,以及它的逆问题——蛋白质设计问题。最近,我们进一步拓展了研究方向,涵盖了RNA和短肽的设计与结构预测,并探索与药物设计相关的课题。总体来讲,我们希望利用最新的人工智能和深度学习技术,结合传统的物理学理论,来解决这些分子生物学以及药物研发相关的基本问题。
黄雨佳:
那您最开始是怎么接触到蛋白质结构预测以及其他生物大分子结构预测这个领域的呢?
张阳:
这是一段有趣的经历。在我的本科和研究生(包括硕士和博士)学习中,我的研究领域是理论物理和粒子物理,主要关注的是物质世界中的基本粒子及其相互作用。后来,在我于中国科学院做博士后期间,偶然读到了欧阳钟灿院士的一篇关于血液中红细胞形状的研究论文,与我之前的理论物理研究方式截然不同,这引起了我的极大兴趣。从那以后,我便开始转向做生物物理领域的研究。
当时,我们课题组使用了一种名为蒙特卡罗模拟的数学算法(Monte Carlo method)。这种方法最初是由物理学家斯坦尼斯瓦夫·马尔钦·乌拉姆(Stanisław Marcin Ulam)和约翰·冯·诺依曼(John von Neumann)在美国洛斯阿拉莫斯国家实验室(Los Alamos National Laboratory)为二战核问题模拟而提出的。蒙特卡罗方法后来被广泛应用于物理学之外的其他领域,包括生物学中蛋白质和RNA的折叠。我当时的研究课题是利用蒙特卡罗方法来研究DNA和RNA分子的弹性属性。
我第一次比较深入接触蛋白质结构预测问题,是在美国加入了杰弗里·什科尔尼克(Jeffrey Skolnick)的实验室做博士后的时候。一开始,我的导师让我基于我之前提出的一种比较新的蒙特卡罗方法,对实验室的蛋白质折叠程序的搜索引擎进行优化。这让我有机会接触到当时蛋白质结构预测领域最前沿的计算机算法。
总的来讲,那几年从理论物理转向生物物理,再到蛋白质结构预测,虽经历了各种酸甜苦辣和艰苦努力,但我觉得这些经历为我后来在蛋白质以及其他生物大分子的折叠和结构预测方面的工作打下了一个较为重要的基础。

新加坡国立大学计算机科学系和生物化学系张阳教授
黄雨佳:
所以为什么科学家会关注用计算机算法来预测蛋白质的结构呢?这个问题为何重要?
张阳:
这个问题之所以重要,是因为蛋白质在生物体生命活动中所扮演的重要角色。我们知道,生命个体中最为重要的两类生物大分子是核酸和蛋白质。其中,核酸(即DNA和RNA分子)作为基因的载体,负责指导生物体合成哪些蛋白质。而生物体内各种生命功能(比如新陈代谢、免疫、催化等)则主要由蛋白质具体执行。蛋白质的功能,即它在细胞中所从事的具体活动,则完全由其三维结构决定。这种结构使蛋白质能参与特定的生物化学反应,并决定它们如何与其他分子进行相互作用。
如果某些蛋白质的功能出现异常,导致疾病的发生,我们需要设计药物来调控这些蛋白质的功能。就像配钥匙的人必须先了解锁孔的形状一样,设计药物的公司必须先要了解这个蛋白质的原子结构,这是蛋白质结构在医学中的一个重要应用。另一方面,如果我们要想详细阐述各种蛋白质在人体内的具体生物学功能,也就是对蛋白质进行功能注解,我们也需要知道它们的三维结构。
传统上,蛋白质分子的原子结构都是通过结构生物学实验来解析的,例如核磁共振、X射线衍射和冷冻电镜等。然而,自然界中的蛋白质种类繁多,实验手段只能解决其中很小的一部分。到2024年10月为止,我们的蛋白质序列数据库已经积累了超过2亿条蛋白质的氨基酸序列,但是只有不到20万条蛋白质序列有实验解析的结构,也就是说只有不到千分之一的已知蛋白质具有实验解析的结构。由于实验解析的耗时和高成本,蛋白质序列和结构的数量差异每天都在增长。因此,若要大规模、系统性地阐述蛋白质的结构和功能,或者进行高通量的药物筛选和设计,实验手段显然远远不够,开发能够精准预测蛋白质结构的计算机算法就显得至关重要。
黄雨佳:
所以用计算机预测蛋白质结构这件事的难点在哪儿呢?
张阳:
这件事的难点主要在两方面。
从数学角度来看,蛋白质就是一条由不同氨基酸串在一起的一维长链,就像一根线串着一串珠子一样。在细胞环境下,它会折叠成一个三维结构,具体折叠成什么样的空间结构,取决于氨基酸的序列排序。从物理学角度,要解决蛋白质折叠和结构预测的问题,我们面临两方面的挑战:第一,理解蛋白质内部原子间的相互作用,即构建一个能精确描述这些相互作用的物理力场;第二,解决高维构象空间的搜索问题。
在物理学力场方面,蛋白质结构的形成依赖于蛋白质内原子之间的各种相互作用,比如氢键、疏水相互作用和范德华力等。这些相互作用在微观尺度上非常复杂,彼此之间相互影响。按照安芬森法则(Anfinsen’s dogma),蛋白质的天然构象是热力学上最稳定的状态,即自由能最小的构象。但我们目前还没有一种能够精准描述这种相互作用的物理学力场,我们也就无法准确计算蛋白质空间构象的最小自由能。这是第一个困难。
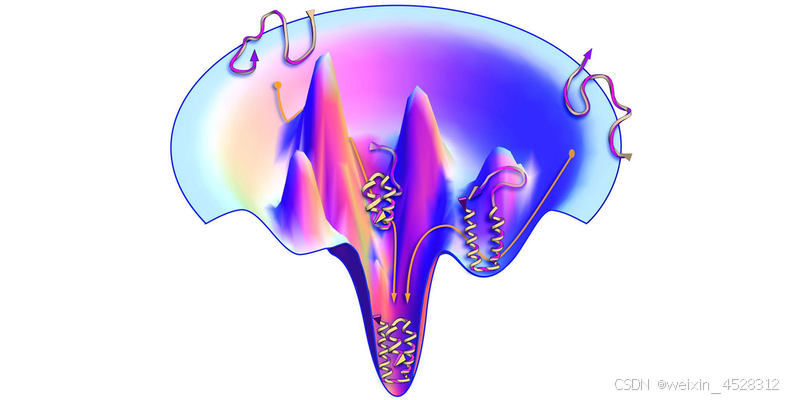
蛋白质结构及其能量景观(landscape)示意图
第二个困难就是高维空间的搜索问题。为什么我一直提到构型搜索呢?因为数学上蛋白质可以有很多种不同的构象。一般来讲,一个普通蛋白质大概含有100至1000个不同的氨基酸。这种长度的链在空间中可能形成的构象的数目非常巨大。50多年前,一个名叫赛勒斯·利文索尔(Cyrus Levinthal)的分子生物学家对此作了估计。他假定每个氨基酸有两个键角——φ和ψ,并假定每个键有3种可能状态。那么对于一个只有100个氨基酸的比较小的蛋白质,它有99个肽键,因此有198个不同的φ和ψ键角,最多就会有种不同构象,约等于
种构象。而且,每个氨基酸的角度状态实际上远不止3种,所以这样小的一个蛋白质的可能构象的数目,可能远远超过我们可观测宇宙中的原子的总数(目前估计大约是
)。
由于构象空间过于庞大,设计一个能够有效搜索并找到自由能最低构象的算法对计算机而言是极具挑战的。我刚才提到的蒙特卡罗方法,是一种通过重点搜索相空间中重要区域来快速折叠蛋白质结构的算法。但是即便如此,它也无法在有限时间内穷尽所有可能相空间的重要构象。
正因为这两方面的挑战,传统基于物理的算法在蛋白质结构预测中面临极大困难。近年来,人工智能和深度学习技术被引入该领域,为解决这些问题提供了新的途径。这个话题我们可以在后面详细讨论,也是这篇文章讨论的一个主要重点。
由于蛋白质结构预测问题的关键性和复杂性,1994年,美国马里兰大学的约翰·莫尔特(John Moult)教授发起了全球蛋白质结构预测比赛(CASP)。CASP每两年举行一次,组织者会发布大约100条蛋白质序列,其中的三维结构要么尚未被解析,要么已解析但未公开。参赛的计算生物学家基于这些序列,运用各自开发的算法来预测其三维结构。竞赛通常持续整个暑假,结束后由独立的科学家团队对结果进行评审,将参赛者的预测结构与实验解析的真实结构进行对比,最接近真实结构的算法被评为最佳。
黄雨佳:
所以这个“最接近”是通过什么标准来评判的呢?
张阳:
这是一个看似简单,实则复杂且非常重要的问题。传统的评判标准是基于均方根偏差(RMSD),即将预测结构与天然结构进行叠加,然后计算每个原子位置的误差,并进行平均。虽然RMSD在概念上易于理解,但它的主要缺陷是对所有原子的误差进行同权平均,导致某些局部区域(如末端或链接区)的较大误差影响整体评分,即使大部分结构预测得很好,RMSD仍可能较差。
为了解决这个问题,我们实验室提出了一种名为TM-score的算法。通过重新设置权重,TM-score更侧重于预测较为精确的区域,因此能够更加准确地反映蛋白质结构预测的整体符合度。从发表论文的统计情况来看,TM-score大概是蛋白质结构预测领域应用最为广泛的评判参数,已经成为这个领域的一个行业标准。
回到刚才的话题,CASP大概上是历史上首次将一个科学问题转化为严格意义上的技术竞赛,因此也被称为“蛋白质结构预测的奥林匹克”。现在,虽然计算机和其他领域都有类似的竞赛,如语言识别、图像识别、网络安全等&
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 846
846

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









