








题目:Integrating Protein Interaction Surface Prediction with a Fragment-Based Drug Design: Automatic Design of New Leads with Fragments on Energy Surfaces
文献来源:https://doi.org/10.1021/acs.jcim.2c01408(JCIM)
代码:https://github.com/colombolab/MLCE
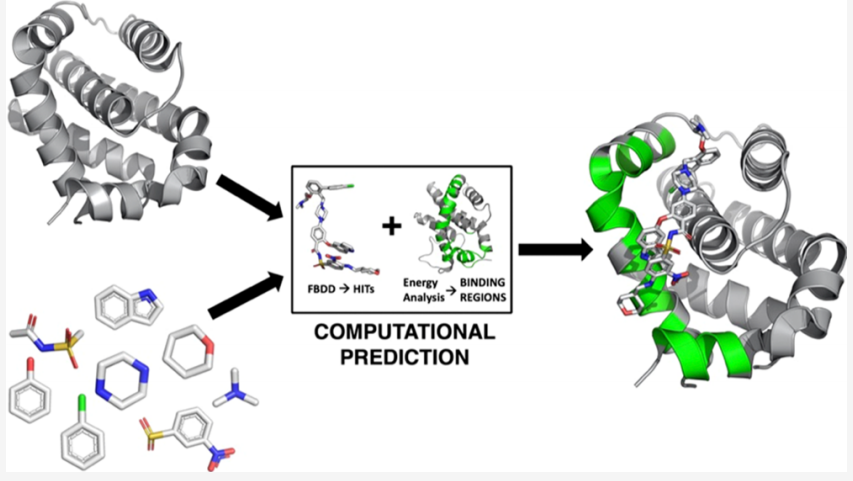
简介:蛋白−蛋白相互作用(PPIs)在过去几年中,由于其在确定病理通路中的关键作用,已成为新疗法开发中的重要药理方法。在这里,作者提出了基于能量表面的片段的设计策略,整合了蛋白质的动态和能量特征的分析,揭示PPIs涉及的亚结构,对接、选择和结合药物样片段从而生成新的PPI抑制剂候选物。具体来说,利用低耦合能量分解矩阵方法,将目标蛋白的结构代表作为基于盲物理的预测潜在蛋白质相互作用表面的输入。预测的相互作用表面被细分为重叠窗口,这些窗口作为模板来指导代表活性药物中典型部分的片段的对接和组合。然后,该方法使用结构多样的、重要的PPI目标作为测试系统进行应用和验证。该方法有助于探索潜在配体的分子多样性空间,并且不需要关于相互作用表面的位置和性质或潜在先导化合物的结构的先验信息。重要的是,从头开始设计中产生的命中分子与实验测试的活性PPI抑制剂具有很高的化学相似性。作者认为他们的方法非常有价值,可以针对困难的目标生成初始线索,以便进一步开发和改进。
主要内容:
-------------------------------------------
来自公众号DrugPython分享
欢迎关注点赞收藏转发!
下次见!


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









