







 本文探讨了蛋白质进化中的趋同和趋异现象,解释了如何通过比较序列和结构来判断进化模式。蛋白质的序列空间和结构空间的概念被介绍,强调了多种序列可以折叠成相同结构的可塑性。HP模型用于简化序列到结构的映射,展示了蛋白质结构的稳定性和对突变的耐受性。
本文探讨了蛋白质进化中的趋同和趋异现象,解释了如何通过比较序列和结构来判断进化模式。蛋白质的序列空间和结构空间的概念被介绍,强调了多种序列可以折叠成相同结构的可塑性。HP模型用于简化序列到结构的映射,展示了蛋白质结构的稳定性和对突变的耐受性。
《Protein Actions Principles and Modeling》-《蛋白质作用原理和建模》
本人能力有限,如果错误欢迎批评指正。
第七章:Proteins Evolve
(蛋白进化)
进化过程可以是趋同的,也可以是趋异的
比较来自不同生物体的现代蛋白质同源物可以帮助我们深入了解它们的祖先。进化可以是趋同的,也可以是趋异的(图7.7)。
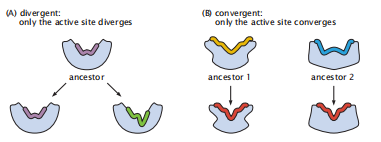
图7.7 趋异进化与趋同进化。(A)趋异:两种蛋白质可以有不同的机制或作用,因为它们与祖先不同(由相似的序列和可能相似的折叠表示)。(B)趋同:两个现代的蛋白质可以具有相似的功能(因为相似的活性位点或结合或调控位点),尽管它们之间的序列同源性较低,这表明它们来自于不同的祖先。
通常在自然界中,两种不同的蛋白质可以发挥相同的生物学功能。例如,胰蛋白酶和弹性蛋白酶是两种不同的丝氨酸蛋白酶。两者都通过水解主链中的肽键来分解其他蛋白质。这两种蛋白质使用相同的催化机制将它们的底物(在这种情况下,这是一个肽键)转化为它们的产物(在这种情况下,是一个断裂的肽键)。趋同进化的例子:两种具有相同生物学功能或机制的蛋白质可以来自于不同的祖先蛋白质。在丝氨酸蛋白酶、天冬氨酸蛋白酶、醛缩酶和拓扑异构酶中已经发现了趋同进化。丝氨酸蛋白酶需要一个催化三联体,一个丝氨酸、组氨酸和天冬氨酸,它们总是排列在相同的空间结构中,来催化肽键的水解,以分解蛋白质。然而,这种催化三联体结构可以出现在完全不同的氨基酸序列和不同的天然折叠中。图7.8 显示了两种碳酸酐酶,其中一个单分子功能是由两个不同的链折叠完成的。
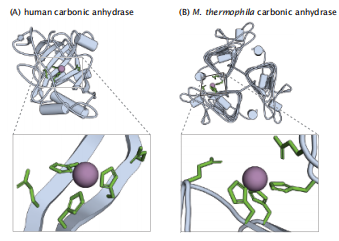
图7.8 趋同进化-两个不同的天然折叠结构具有相同的功能。(A)人碳酸酐酶具有α/β卷折叠结构,而(B)来自嗜热甲烷菌的碳酸酐酶具有β-螺旋拓扑结构。嗜热支原体蛋白与任何碳酸酐酶都没有显著的序列同源性,代表了一类独特的碳酸酐酶。
相反,在趋异进化中,来自于同一个祖先的蛋白质可以具有两种非常不同的蛋白质功能。图7.9 显示了不同的蛋白质折叠可以具有多少种功能。例如,tim-bebld蛋白质结构家族的成员具有16种不同的已知功能。图中显示,在一个具有229个结构的数据库中,175个蛋白质具有单一已知功能,38个具有两个已知功能,少数例具有多个功能。有时,蛋白质的功能是适应性的:当在一种构象时执行一种功能的蛋白质,在不同构象时可以执行不同的功能。例如,OB折叠与不同的单链DNA片段结合,但蛋白质可以改变构象来识别不同DNA分子的不同结合特征。
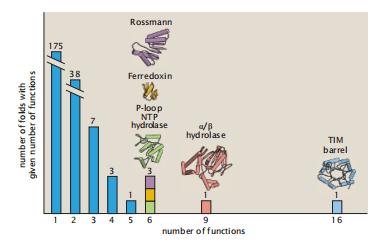
图7.9 有多少个蛋白质折叠结构有多少种不同的功能?TIM桶是一个有16个已知功能的折叠结构(最右图)。有175个结构,每个可以执行一个功能(最左)。此外,根据铁氧还蛋白、罗斯曼折叠和β-turn水解酶折叠结构,每种结构都有6种功能。
那么如何确定趋同进化还是趋异进化呢(参见图7.7)?比较两种同源蛋白。这可以通过序列匹配和结构匹配,以及基因本体论术语的比较来确定。如果蛋白质有相似的序列和不同的机制,那么它们就会有所不同。如果蛋白质有不同的序列和相似的机制,那么它们就是趋同的。当然,蛋白质可以在漫长的进化过程中产生足够的差异,因此没有任何属性的相似之处——无论是序列、结构或功能。当蛋白关系太遥远时,你就无法推断出它们之间的进化亲缘关系。
系统发育树显示了生物体或序列之间的进化关系
在达尔文时代,进化缘关系是通过比较两种生物之间的形态(解剖结构)来建立的,比如它们的骨骼、牙齿、翅膀或叶子。然而,自20世纪60年代以来,越来越多的基于生物分子序列的定量方法来构建进化树。
图7.1 展示的是一个进化树。每个节点代表一个生物体,无论是已知的还是未知的。节点之间的边(称为分支)具有不同的长度。分支的长度代表了两个生物体之间的相对进化距离。分支长度越长,两个生物体之间的距离就越远。要构建这样的树,您首先需要定义一个度量标准来确定两个物种、分子或序列之间的进化距离。从Box7.1中回想一下,Hamming距离就是这样一个度量标准。Hamming距离越大,两个序列之间的差异就越大。
第8章描述了如何根据在进化谱系中发现的序列来构建蛋白质的进化树。首先使用对齐方法排列不同的氨基酸序列。然后你计算序列对之间的序列相似性得分。然后,通过绘制每条边的长度与相应的序列相似度得分成比例来构造该树。像家谱一样,进化树可以表达不同生物体之间的历史和关系。一个重要的问题是氨基酸序列是如何与天然结构相关联的?
-许多不同的序列可以折叠成相同的天然结构
为了解决序列-结构关系的问题,我们从序列空间的概念开始。我们可以用许多不同的方式将这20种不同的氨基酸串在一起。所有可能包含N个氨基酸的氨基酸序列的集合称为长度为N的蛋白质序列空间。任何一个特定的氨基酸都被表示为序列空间中的一个单个点。它被称为空间-我们可以使用网格点的N维数学坐标系来描述它(图7.10)。沿着第一个坐标轴,叫它x1,你有20个点,每个点代表20个氨基酸中的一个。沿着第二个轴x2,还有20个点代表氨基酸。所有的N个轴,直到xN,都用这样的点来标记。轴x1表示序列中出现在位置1处的氨基酸;轴x2表示出现在位置2的氨基酸,以此类推。图7.10显示了序列空间中的一个点如何代表一个氨基酸,例如(x1,x2,x3,…)=(Ala,Gly、Trp,…)。
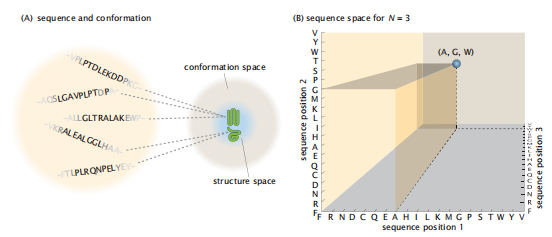
7.10 什么是序列空间?它是如何映射到结构空间的?(A)不同的序列可以折叠成不同的结构(绿色图标)。许多不同的序列可以折叠成一个特定的天然结构。(B)序列空间。每个轴上有20个氨基酸的编号。轴的数目是链长n。所以,在这个N维空间中的一个点代表一个氨基酸序列。这里显示的是在这样一个空间中的一个三残基链的序列AGW。
序列空间有多大?如果链中的每个氨基酸都是从M-字母字母表中选择的(M = 20表示天然氨基酸)中选择的,那么在每个N轴上就会有M个可能的点。因此,序列空间有MN网格点,表示该链长度的所有可能的不同序列。例如,考虑一个链长为N = 100个氨基酸的蛋白质,来自M=20个氨基酸。该长度的不同序列数将为20100= 1.3×10130。序列空间的概念有助于对进化的理解。在进化过程中,蛋白质受到突变变化,如一种氨基酸交换另一种氨基酸或氨基酸的丢失或获得。我们可以将进化过程中的突变变化看作是在序列空间中从一个网格点到另一个网格点的跳跃或扩散。
一个蛋白质也有一个构象空间,即一个给定长度的链分子的所有可能构象的集合(见图7.10)。构象空间中的每个点代表一个链的构象。对于具有N个残基的蛋白质,每个残基包含两个可旋转键(φ和ψ角),每个残基具有z旋转构象,对于大N,构象空间的大小估计为z2N。构象的空间进一步受到蛋白质类性质的限制,如高堆积密度、疏水核的形成和二级结构的倾向。类天然的致密构象只构成构象空间的一小部分。大多数构象空间是大量的无序和变性构象。
HP模型对序列结构映射可以提供一些看法
蛋白质的序列和结构之间的关系是什么?我们从简化字母表模型的研究中得到了一些见解,如第3章的HP模型。对于在二维正方形晶格上有N= 16个单体的HP链(每一步有z=3个可达方向),序列空间包含216个= 65,536个序列(使用M = 2表示两种残基类型,H和P),所有构象的(不包括那些与对称相关的构象)空间有802,075个构象。致密构象的数量要小得多:例如,16长度的序列只有69个完全致密构象适合4×4晶格。因此,你可以通过计算机使用晶格上的HP蛋白来研究所有可能序列的所有可能构象。这些简化模型可以捕获序列结构映射的一些基本规律。
HP建模显示,将序列映射到本机结构是多对一的(参见图7.10A)。也就是说,许多不同的氨基酸序列可能会折叠成相同的天然结构。这些信息对于理解蛋白质的可塑性或对突变的鲁棒性是有用的。如果你用不同的氨基酸替换链序列中一个位置的氨基酸,这就被称为单位点突变。天然蛋白质被称为野生型蛋白质,突变版本被称为突变蛋白。一个单位点突变对应于序列空间中从一个点到附近点的一步,就像青蛙在池塘里从一个百合叶跳到下一个点一样。通常,从一个序列S改变到另一个序列S'导致不改变的最低能量结构的蛋白质折叠:两个序列折叠到相同的原生结构N.所有折叠到一个给定的原生结构的序列集合被称为中性集或收敛集(neutral set 或者 convergence set)。
如果一个蛋白质结构有一个较大的收敛集,则称为可设计的蛋白质结构。也就是说,如果许多不同的序列折叠成一个蛋白质结构,那么说明蛋白质结构是可设计的。一个可设计的结构对氨基酸的变化是非常稳定的:各种突变不会导致蛋白质的天然结构的变化。HP模型表明,蛋白质结构通常应该对单一氨基酸的变化具有高度的耐受性。
在20世纪70年代之前,人们认为一种蛋白质不能在不破坏其结构的情况下耐受许多突变。蛋白质的天然结构被认为是脆弱的,也就是说,相当刚性和不可改变的。在那些日子里,对蛋白质突变的研究很少见,而且突变只存在蛋白质内的随机位置。1978年,在改变蛋白质科学的工作中, Michael Smith发明了定点或单位点或点突变,这是一种更有针对性的方法,可以用蛋白质序列中的一种氨基酸取代另一种氨基酸。为此,他共同获得了1993年的诺贝尔奖。现在有了大量的序列。例如,仅已知的激酶序列就有超过5万个序列。研究序列可以告诉你很多关于哪些替换是可以合适的。
从几十年来对靶向突变的广泛研究中,现在可以清楚地看到,蛋白质的天然结构是具有可塑性和很稳定的-对点突变具有高度耐受性。也就是说,因为蛋白质具有很大的收敛集所以天然结构可以被编码到许多不同的序列中。例如,在T4溶菌酶]和ROP蛋白中,整个疏水核已经被其他疏水残基所取代。
如何用不同的序列来编码一个单一的天然结构呢?框7.2显示了两个不同的HP序列,它们可以折叠成相同的天然序列。
==================================================
BOX 7.2 A 给定的天然折叠结构可以由不同的序列编码
在HP模型中,原生状态由HH接触的最大数量决定。在图7.11中,我们可以看到两个不同的HP序列,它们的原生结构是相同的。在这里,两个不同的序列折叠成相同的天然结构,即使天然HH接触的数量相差1。一般来说,对于更长的链和更细粒度的模型,一个单一的原生结构可以由大量不同的序列进行编码。
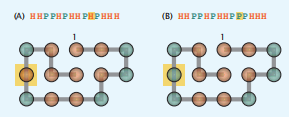
图7.11这两个不同的HP序列(A)和(B)编码相同的原生折叠。序列的不同在于黄色框表示的一个位置。橙色的球体代表疏水(H)残基,绿色的球体代表极性(P)残基
==================================================
图7.12说明了许多不同的序列可以编码相同的天然折叠。它显示了逆折叠问题或蛋白质设计问题。在这类任务中,我们确实需要给出一个特别致密的目标蛋白结构。如果我们想要设计一个序列,其特定的结构是最稳定的构象。要设计它,首先将链主干绘制到您想要的本地配置中的晶格上。在这个设计过程的这个阶段,每个氨基酸珠还没有一个H或p的身份,它只是一个空白链。然后根据一些配方在每个残留位置上“涂”H或P来设计序列。通过这种方式,您将采用一个目标结构,并将设计出一个可以折叠起来的序列。
在HP模型中,设计方法相对简单。对于位于结构核心的氨基酸位置,你可以分配H类型的单体,以制造一个疏水核心。对于蛋白质结构表面的位点,无论分配H还是P并不重要,因为表面相互作用对(B)的稳定性没有太大影响。从本质上说,通过设计一个核心有H单体,表面有P单体的序列,是做创造稳定疏水核心的积极设计。而且,把P单体放在表面上是在做一些消极的设计,以确保其他(错误的)链折叠不太稳定。当然,设计真正的蛋白质也需要考虑其他因素。
-------------------------------------------
欢迎点赞收藏转发!
下次见!


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









