







文献介绍

文献题目: Spatial multi-omics at subcellular resolution via high-throughput in situ pairwise sequencing
研究团队: 曹罡(深圳理工大学)、戴金霞(华中农业大学)
发表时间: 2024-05-14
发表期刊: Nature Biomedical Engineering
影响因子: 28.1
DOI: 10.1038/s41551-024-01205-7
摘要
空间多组学技术有助于发现细胞功能和疾病机制的新见解。在这里,我们报告了多组学原位成对测序 (MiP-seq) 的开发和应用,这是一种以亚细胞分辨率同时检测 DNAs、RNAs、蛋白质和生物分子的方法。与其他原位测序方法相比,MiP-seq 增强了解码能力,降低了测序和成像成本,同时保持了检测基因突变、等位基因特异性表达和 RNA 修饰的效率。MiP-seq 可以整合体内钙成像和拉曼成像,这使我们能够生成小鼠脑组织的空间多组学图谱,并将基因表达与神经元活动和细胞生化指纹相关联。我们还报告了一种连续稀释策略,用于在原位测序过程中解决光学拥挤信号。高通量原位成对测序可以促进组织分子和功能图谱的多维分析。
前言
多细胞生物优雅地协调特定细胞中适当基因的共转录。这些空间转录谱控制不同细胞的功能,以完成复杂的生理任务。全面空间组学的描述将为理解特定细胞的分子功能铺平道路,并通过例如在癌症治疗前表征免疫微环境来极大地推进精准诊断。
最近,最先进的空间转录组学方法已被开发用于高度多重 RNA 原位检测。一般来说,这些空间转录组学方法是基于三种策略开发的:多重荧光原位杂交(FISH)(如 SeqFISH 和 MERFISH)、原位测序(如 ISS、FISSEQ、STARmap、INSTA-seq 和 ExSeq) 、原位捕获与高通量测序相结合(如 Slide-seq、HDST 和 DBiT-seq)。然而,空间组学技术仍处于起步阶段。例如,大多数方法只能破译一种生物分子的空间信息,多种生物分子的原位联合检测仍然具有挑战性。此外,由于大多数基于原位测序和基于 FISH 的方法仅具有 (4 代表四个荧光基团,N 代表测序或杂交轮数)解码能力,因此需要多轮测序才能实现高通量空间组学。需要提高捕获效率、通量、细胞分辨率和光学拥挤以及降低这些实验成本的方法。在这里,我们报告了一种高通量靶向原位测序方法,多组学原位成对测序(multi-omics in situ pairwise sequencing, MiP-seq),的开发,能够以亚细胞分辨率有效检测脑组织中的多重 DNAs、RNAs、蛋白质和生物分子。
研究结果
1. MiP-seq 在生物分子空间分析中的应用
MiP-seq 对 DNA 和 RNA 的原位检测是通过针对核酸的直接锁式探针实现的,而蛋白质和生物分子的检测是通过检测抗体偶联核酸的锁式探针实现的(Fig. 1a)。首先,我们将 MiP-seq 应用于空间 RNA 分析,该过程涉及锁式探针杂交、原位滚环扩增 (RCA) 和基于连接的双条形码测序(Fig. 1b)。值得注意的是,每个锁式探针包含两个条形码(B1 和 B2),这增加了其信号解码能力。根据之前的研究,我们比较了不同的连接酶(Supplementary Fig. 1)并选择 SplintR 来连接 RCA 模板,这提高了 RCA 模板连接效率。这种连接方法依赖于精确匹配的碱基配对,从而可以通过 MiP-seq 检测目标 RNA 中的单核苷酸多态性 (SNP)。由于锁式探针和起始引物(IP)的目标依赖性连接与目标 RNA 和锁式探针互补,RCA 与目标一起牢固地在原位进行(Fig. 1b)。为了进行高通量检测,通过连接和信号解码对原位滚环扩增产物(RCPs)进行多轮测序(Fig. 1b)。
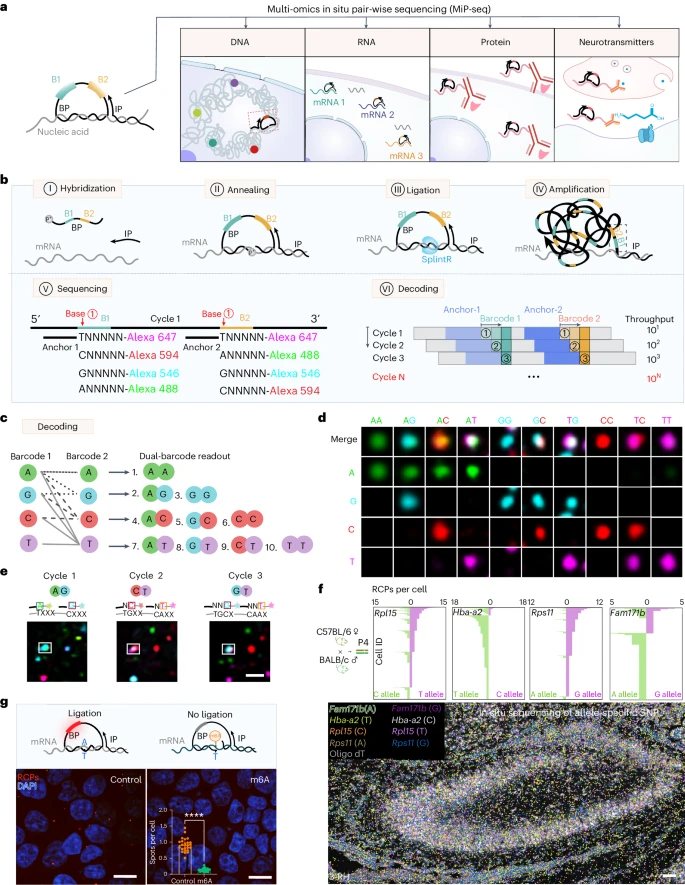
a. MiP-seq 在空间组学分析中的应用图。
b. 用于 RNA 原位检测的 MiP-seq 流程图。I. 双条形码引物 (BP) 和起始引物 (IP) 与目标 RNA 杂交。II. BP 和 IP 与目标 mRNA 退火形成锁式结构。III. BP 通过 RNA 依赖性 DNA 连接酶 SplintR 环化形成 RCA 模板。IV. 圆形 BP 的 RCA 形成滚环产物。V. 双锚定引物在双条形码序列旁边杂交。随后,通过连接测序,荧光标记的查询探针对条形码序列进行查询。VI. 每轮测序解码 10 个双条形码读数,N 轮后可查询
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 633
633

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











