






一、参考论文和博文
统计量面面观之基因表达的组织特异性 - 知乎 (zhihu.com)

二、如何利用K-means聚类来确定划分TSgenes的Tau值
(一)方法1:目前Tau=0.85是根据已有文献来的,现在我们第一步根据轮廓系数计算K(2-100)最佳值,第二步根据这个最佳值的全基因聚类看Tau=1的基因分布于在哪些cluster中。分析cluster基因的Tau值分布区间,取最小的Tau值作为划分TSgenes的值。目前Tau=0.85是根据已有文献来的,现在我们第一步根据轮廓系数计算K(2-100)最佳值,第二步根据这个最佳K值的全基因聚类看Tau=1的基因分布于在哪些cluster中。分析cluster基因的Tau值分布区间,取最小的Tau值作为划分TSgenes的值。
import numpy as np
from sklearn.cluster import KMeans
from sklearn.metrics import silhouette_score
import pandas as pd
# 从文件中读取数据
data = pd.read_csv("/home/share_data1/luchh/new_TS/kmeanstest/Pto_TS_fpkm_new.txt", sep='\t', index_col=0)
# 转换数据框为numpy数组
X = data.values
# 初始化变量以存储轮廓系数的信息和最佳聚类结果
best_k = 0
best_silhouette_score = -1
best_labels = None
# 初始化变量以存储轮廓系数的信息
results = []
# 选择一系列可能的K值进行尝试
for k in range(2, 100): # 从2到99尝试不同的K值
kmeans = KMeans(n_clusters=k, random_state=0)
labels = kmeans.fit_predict(X)
silhouette_avg = silhouette_score(X, labels)
results.append({'K值': k, '轮廓系数': silhouette_avg})
# 输出当前K值和轮廓系数
print(f"K={k}, 轮廓系数={silhouette_avg}")
# 如果当前K值的轮廓系数更好,更新最佳K值和轮廓系数,并记录最佳的聚类结果
if silhouette_avg > best_silhouette_score:
best_k = k
best_silhouette_score = silhouette_avg
best_labels = labels
# 创建一个数据框来总结结果
results_df = pd.DataFrame(results)
# 保存结果到CSV文件
results_df.to_csv("kmeans_results.csv", index=False)
# 保存最佳聚类结果的标签到CSV文件
best_result_df = pd.DataFrame({'K值': best_k, '轮廓系数': best_silhouette_score}, index=[0])
best_result_df.to_csv("best_kmeans_result.csv", index=False)
# 保存最佳聚类结果的标签到CSV文件
best_labels_df = pd.DataFrame({'GeneID': data.index, 'ClusterLabel': best_labels})
best_labels_df.to_csv("best_kmeans_labels.csv", index=False)
从结果可以看到,所有数据只要一分类轮廓系数就下降,最好的是K=2,所以无法用这样的方法选择τ值。

(二)方法2:第一步:根据Tau值(0.8、0.85、0.90、0.95…)获取Tsgenes,可以做一个Tau值与Tsgenes数目的对应表格,选取Tsgenes数目一样的tau值;第二步:对TSgenes进行K-means(>组织数)聚类,根据轮廓系数计算K最佳值;第三步:最佳K值=组织数,Tau=1基因全部在不同cluster,这样有些基因虽然Tau值相对较低,但是也能较好的分为对应组织数量,说明与RootTSgenes(Tau=1)在同一cluster的基因也应该被认为是RootTSgenes。
第一步:
(1)固定tau最低值为0.8,max值初始=min值,每一次max+0.001
import pandas as pd
# 从文件中读取数据
data = pd.read_csv("/home/share_data1/luchh/new_TS/kmeanstest/test/tau.txt", sep='\t', usecols=[0], header=None, names=["Value"])
# 固定的 min_value 值
min_value = 0.8
results = [] # 存储统计结果的列表
max_value = min_value # 初始 max_value 等于 min_value
while max_value <= data["Value"].max(): # 以最大数据点为限制条件
count = data[(data["Value"] >= min_value) & (data["Value"] <= max_value)]["Value"].count()
results.append((min_value, max_value, count))
max_value += 0.001
# 创建一个数据框来存储统计结果
result_df = pd.DataFrame(results, columns=["Min_Value", "Max_Value", "Count"])
# 将结果保存为CSV文件
result_df.to_csv("result.csv", index=False)
gene_count结果都不一样,所以无法减少tau值步长

(2)固定min和max间隔大小0.05,min每一次加0.01
import pandas as pd
# 从文件中读取数据
data = pd.read_csv("/home/share_data1/luchh/new_TS/kmeanstest/test/tau.txt", sep='\t', usecols=[0], header=None, names=["Value"])
# 初始的 min_value 和 step 值
min_value = 0.8
step = 0.01
results = [] # 存储统计结果的列表
while min_value <= data["Value"].max(): # 以最大数据点为限制条件
max_value = min_value + 0.05 # max_value 始终是 min_value + 0.05
count = data[(data["Value"] >= min_value) & (data["Value"] <= max_value)]["Value"].count()
results.append((min_value, max_value, count))
min_value += step
# 创建一个数据框来存储统计结果
result_df = pd.DataFrame(results, columns=["Min_Value", "Max_Value", "Count"])
# 将结果保存为CSV文件
result_df.to_csv("result.csv", index=False)
结果也是gene_counts数都不一样。
第二步,从Tau=0.8开始,每次增加0.01计算TSgene的轮廓系数,best-K最好是14,加入长短枝数据,K值永远是2,不要加进去,
import pandas as pd
from sklearn.cluster import KMeans
from sklearn.metrics import silhouette_score
# 读取txt文件
data = pd.read_csv("/home/share_data1/luchh/new_TS/kmeanstest/TauandbeatK/nonshortlong/Pto_TS_fpkm_nonshortlong_Tau.txt", sep='\t', index_col=0)
# 初始化一个空的DataFrame来存储结果
result_df = pd.DataFrame(columns=['min_tau', 'best_K'])
# 初始的 min_tau 值为数据中的最大值
min_tau = data['Tau'].max()
while min_tau >= 0.8:
# 筛选"Tau"列中数值大于等于 min_tau 的数据
filtered_data = data[data['Tau'] >= min_tau]
# 移除 "Tau" 列
filtered_data = filtered_data.drop(columns=['Tau'])
best_silhouette_score = -1
best_K = 0
possible_K_values = range(2, 20)
for K in possible_K_values:
kmeans = KMeans(n_clusters=K, random_state=0)
cluster_labels = kmeans.fit_predict(filtered_data)
silhouette_avg = silhouette_score(filtered_data, cluster_labels)
if silhouette_avg > best_silhouette_score:
best_silhouette_score = silhouette_avg
best_K = K
# 将结果添加到 result_df
result_df = result_df.append({'min_tau': min_tau, 'best_K': best_K}, ignore_index=True)
# 减去0.01的 min_tau 值
min_tau -= 0.001
# 保存结果到 'best_K_values.csv'
result_df.to_csv('best_K_values.csv', index=False)
结果如下,并没有best-K=14的tau值
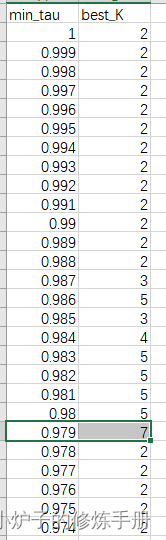
选择kmeans最大的0.979作为TSgenes分类标准。筛选tau=1的基因发现,root基因和很多其他组织基因没有区分开来,在一个cluster中,所以无法用kmeans来判断tau值取舍。
所以后续应该怎么判断tau的阈值呢?以下是初步设想。
三、如何利用支持向量机来确定划分TSgenes的Tau值
读取基因表达数据txt文件,根据最后一列Tau值大小进行分类,大于0.8的为组织特异性性基因记为“1”,小于0.8的为非组特异性基因记为“0”,筛选所有组织特异性基因的表达数据和随机同等数量的非组织特异性基因的表达数据作为数据集,分为训练集和测试集,创建一个支持向量机分类器,训练分类器,进行十折验证,预测概率,计算 ROC 曲线和 AUC,绘制 ROC 曲线。















 5993
5993











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









