






一、gff文件准备
1、准备基因gff文件
从数据库下载
2、excel整理获得输入gff文件
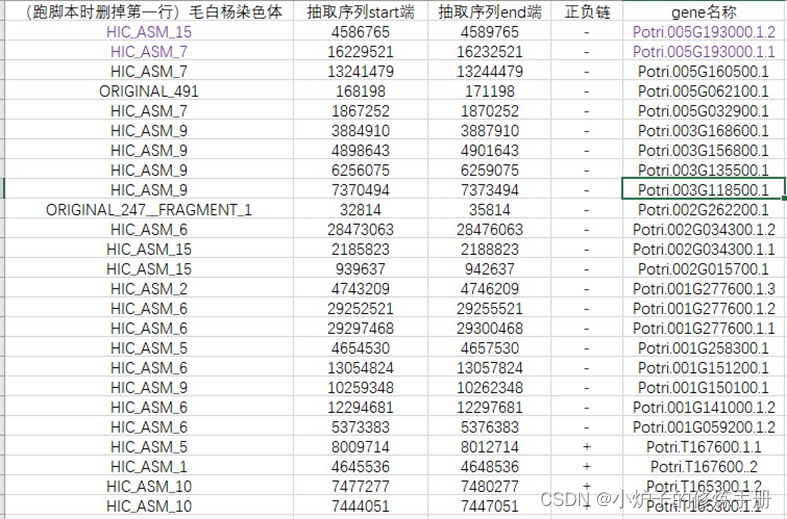
(1)计算正负链:比对结果end-start>0,为正链,反之负链。
(2)在正链中,将小数值减2000(向前2000bp)为一列,向后加200(向后200bp)为一列,并且删掉原来的数值;在负链中,将大数值加2000,小数值减200,得到的是启动子区域的位置。(启动子大小自己确定)
(3)这样的处理-2000可能会出现负数,将负数改成1就好。
(4)最终格式如下:抽取序列时,都是小数值在前,保存为gff.txt
二、从基因组抽取启动子序列
利用script(Extract_seq_previ_termi_v4.py),可以根据位置信息提取基因组的相关序列。
经过excel整理后,其实运行代码命令如下:
python2 Extract_seq_previ_termi_v2.py genome.fa gff.txt 0 0#!/usr/bin/env python
from __future__ import division
print '''
Usage: python Extract_seq_previ.py genome.fa gff previ_length termi_length
gff format:NC_001147.6 159548 160594 - genename
length: bp
'''
import sys,re
IN1=open(sys.argv[1],'r') # genome file
IN2=open(sys.argv[2],'r') # glimmer file format:NC_001147.6 159548 160594 -
kb1=int(sys.argv[3].strip()) # previous N bp
kb2=int(sys.argv[4].strip()) #termi N bp
OUT=open("fa.out",'w')
def Max(i,j):
if int(i)>=int(j):
return int(j)
else:
return int(i)
def Seqin(fa):
seq_name=[]
seqs=[]
each_seq=""
for eachline in fa:
eachline=eachline.rstrip()
if eachline.startswith(">"):
seq_name.append(eachline.strip(">"))
if each_seq:
seqs.append(each_seq)
each_seq=""
else:
each_seq+=eachline
seqs.append(each_seq)
return seq_name,seqs
def SeqConvert(seq):
Convert={"A":"T","C":"G","T":"A","G":"C","a":"t","t":'a',"c":'g',"g":"c","N":"N","n":"n"}
return ''.join(map(lambda x: Convert[x],seq))[::-1]
seq_name=[]
seqs=[]
seq=""
seq_name,seqs=Seqin(IN1)
#for i in seq_name:
# print ">"+i+'\n'+str(len(seq[seq_name.index(i)]))
split=[]
for eachline in IN2:
split=eachline.rstrip().split('\t')
try:
if split[3]=="-":
seq=seqs[seq_name.index(split[0])][(int(split[1])-Max(int(split[1]),kb2)):(int(split[1])-1)].lower()+seqs[seq_name.index(split[0])][(int(split[1])-1):(int(split[2]))].upper()+seqs[seq_name.index(split[0])][(int(split[2])):(int(split[2])+kb1)].lower()
seq=SeqConvert(seq)
else:
seq=seqs[seq_name.index(split[0])][(int(split[1])-Max(int(split[1]),kb1)):(int(split[1])-1)].lower()+seqs[seq_name.index(split[0])][(int(split[1])-1):(int(split[2]))].upper()+seqs[seq_name.index(split[0])][int(split[2]):(int(split[2])+kb2)].lower()
OUT.write(">"+'_'.join([split[4],str(kb1),str(kb2)])+'\n'+seq+'\n')
except IndexError:
print "pass"
IN1.close()
IN2.close()
OUT.close()
三、从fasta文件,根据ID抽取序列
只选择其中几个启动子,可以用script (extract_fasta_by_id.py)根据基因的名称提取启动子序列
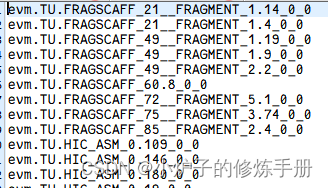
(1)准备基因ID_list
注意ID与fasta的分时相同
(2)根据ID_list抽取对应fasta序列
python2 extract_fasta_by_id.py id.txt(需要抽取的Gene名称) genome.fa(序列文件) seq.fa(结果) #! /usr/bin/env python
import sys,re
IN1=open(sys.argv[1],'r')
IN2=open(sys.argv[2],'r')
OUT=open(sys.argv[3],'w')
lst=[]
k=0
for eachline in IN1:
eachline=eachline.rstrip()
lst.append(eachline)
for eachline in IN2:
m=re.search("(>(\S+))",eachline)
if m:
name=m.group(2)
if name in lst:
k=1
OUT.write("%s%s\n"%(">",m.group(2)))
else:k=0
else:
if k==1:
OUT.write(eachline)
IN1.close()
IN2.close()
OUT.close()
import sys
from Bio import SeqIO
from Bio.Seq import Seq
from collections import defaultdict
# 解析GFF文件,提取基因位置信息
def parse_gff(gff_file, feature_type='gene'):
genes = defaultdict(dict)
with open(gff_file, 'r') as f:
for line in f:
if line.startswith('#'): # 跳过注释行
continue
fields = line.strip().split('\t')
seqid, source, feature, start, end, score, strand, frame, attributes = fields
if feature == feature_type: # 只提取特定类型的注释,如'gene'
attributes_dict = {key_value.split('=')[0]: key_value.split('=')[1] for key_value in attributes.split(';')}
gene_id = attributes_dict.get('ID', None)
if gene_id:
genes[seqid][gene_id] = {'start': int(start), 'end': int(end), 'strand': strand}
return genes
# 从FASTA文件中提取指定基因组区域
def extract_sequence(genome_file, seqid, start, end):
genome = SeqIO.to_dict(SeqIO.parse(genome_file, 'fasta'))
sequence = genome.get(seqid, None)
if sequence:
return sequence.seq[start-1:end]
return None
# 提取启动子区域
def extract_promoter_sequence(genome_file, gff_file, gene_list_file, promoter_length, feature_type='gene'):
# 读取GFF文件中的基因注释
genes = parse_gff(gff_file, feature_type)
# 读取基因ID列表
with open(gene_list_file, 'r') as f:
gene_list = [line.strip() for line in f.readlines()]
promoters = []
# 提取启动子序列
for seqid, gene_info in genes.items():
for gene_id, gene_data in gene_info.items():
if gene_id in gene_list:
start = gene_data['start']
end = gene_data['end']
strand = gene_data['strand']
# 启动子区域长度从基因的起始位置计算
if strand == '+':
promoter_start = max(1, start - promoter_length) # 防止负值
promoter_end = start - 1
else:
promoter_start = end + 1
promoter_end = end + promoter_length
promoter_seq = extract_sequence(genome_file, seqid, promoter_start, promoter_end)
if promoter_seq:
promoters.append(f">{gene_id}_promoter\n{promoter_seq}")
return promoters
# 写入FASTA文件
def write_fasta(promoters, output_file):
with open(output_file, 'w') as f:
for promoter in promoters:
f.write(promoter + '\n')
# 主函数
def main():
if len(sys.argv) != 6:
print("用法: python script.py <gff_file> <genome_file> <gene_list_file> <promoter_length> <output_file>")
sys.exit(1)
# 从命令行参数中获取文件路径和长度
gff_file = sys.argv[1]
genome_file = sys.argv[2]
gene_list_file = sys.argv[3]
promoter_length = int(sys.argv[4])
output_file = sys.argv[5]
promoters = extract_promoter_sequence(genome_file, gff_file, gene_list_file, promoter_length)
write_fasta(promoters, output_file)
print(f"FASTA文件已保存到{output_file}")
if __name__ == '__main__':
main()

















 865
865

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









