







1.前言
书接上回,上篇分享了作者做的基于bulkRNA的细胞分类评估工具CellNet及其更新版PACNet,现在分享的是单细胞数据的细胞分类评估工具singleCellNet,该工具与CellNet是同时期开发的。
singleCellNet:https://github.com/CahanLab/singleCellNet
单细胞RNA测序(scRNA-seq)技术正迅速成为生成器官、组织和生物体细胞图谱的关键工具。它有助于定义发展阶段、调控因素,以及了解年龄、疾病或遗传变异如何影响细胞组成和状态。scRNA-seq中最耗时的环节之一是“细胞分型”——确定每个细胞的身份,这通常需要额外的实验步骤,如原位定位或功能评估。因此,迫切需要一种更快速、更精确的定量分型方法。
一种解决方案是将待分析的scRNA-seq数据与已鉴定细胞类型的现有数据集进行整合。已有多种方法提出用于整合scRNA-seq数据集,以提高分析的统计效力。然而,这些方法通常需要两个数据集中至少有一种共同的丰富细胞类型,且它们并未提供与参考数据集相比对查询细胞类型进行定量分类的方法。
特点:
• SingleCellNet(SCN)可对scRNA-seq数据进行定量分类
• SCN可以跨平台、跨物种应用
• SCN可以评估细胞命运工程实验的保真度
• SCN 提供 12 个即用型公共参考数据集
2.singleCellNet简介
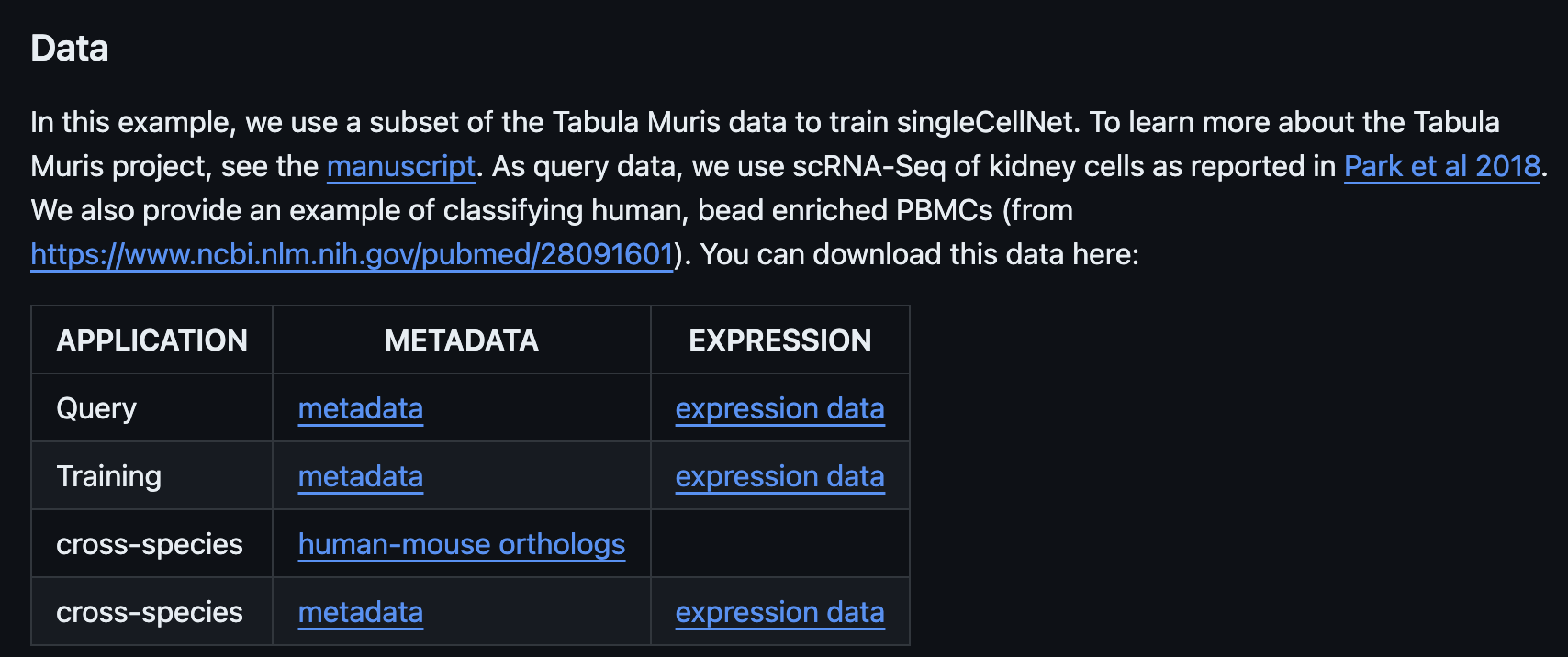
整个框架及思路与bulk的处理大同小异的,且特色还是多物种和多分组,这里作者依旧提供了PBMC的测试数据:
下载R包:
install.packages("devtools")
devtools::install_github("pcahan1/singleCellNet")
library(singleCellNet)
下载数据:
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/sampTab_Park_MouseKidney_062118.rda", "sampTab_Park_MouseKidney_062118.rda")
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/expMatrix_Park_MouseKidney_Oct_12_2018.rda", "expMatrix_Park_MouseKidney_Oct_12_2018.rda")
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/expMatrix_TM_Raw_Oct_12_2018.rda", "expMatrix_TM_Raw_Oct_12_2018.rda")
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/sampTab_TM_053018.rda", "sampTab_TM_053018.rda")
## For cross-species analyis:
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/human_mouse_genes_Jul_24_2018.rda", "human_mouse_genes_Jul_24_2018.rda")
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/6k_beadpurfied_raw.rda", "6k_beadpurfied_raw.rda")
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/stDat_beads_mar22.rda", "stDat_beads_mar22.rda")
## To demonstrate how to integrate loom files to SCN
download.file("https://s3.amazonaws.com/cnobjects/singleCellNet/examples/pbmc_6k.loom", "pbmc_6k.loom")
3.singleCellNet demo
加载查询数据:
stPark = utils_loadObject("sampTab_Park_MouseKidney_062118.rda")
expPark = utils_loadObject("expMatrix_Park_MouseKidney_Oct_12_2018.rda")
dim(expPark)
# [1] 16272 43745
genesPark = rownames(expPark)
rm(expPark)
gc()
加载测试数据:
expTMraw = utils_loadObject("expMatrix_TM_Raw_Oct_12_2018.rda")
dim(expTMraw)
# [1] 23433 24936
stTM = utils_loadObject("sampTab_TM_053018.rda")
dim(stTM)
# [1] 24936 17
stTM<-droplevels(stTM)
拆分训练集和测试集:
## 交集共有基因
commonGenes = intersect(rownames(expTMraw), genesPark)
length(commonGenes)
# [1] 13831
expTMraw = expTMraw[commonGenes,]
## 拆分数据
set.seed(100) #can be any random seed number
stList = splitCommon(sampTab=stTM, ncells=100, dLevel="newAnn")
stTrain = stList[[1]]
expTrain = expTMraw[,rownames(stTrain)]
训练分类器:
system.time(class_info<-scn_train(stTrain = stTrain, expTrain = expTrain, nTopGenes = 10, nRand = 70, nTrees = 1000, nTopGenePairs = 25, dLevel = "newAnn", colName_samp = "cell"))
# user system elapsed
# 476.839 25.809 503.351
提取数据进行分类器评估:
#validate data
stTestList = splitCommon(sampTab=stList[[2]], ncells=100, dLevel="newAnn") #normalize validation data so that the assessment is as fair as possible
stTest = stTestList[[1]]
expTest = expTMraw[commonGenes,rownames(stTest)]
#predict
classRes_val_all = scn_predict(cnProc=class_info[['cnProc']], expDat=expTest, nrand = 50)
4.评估结果
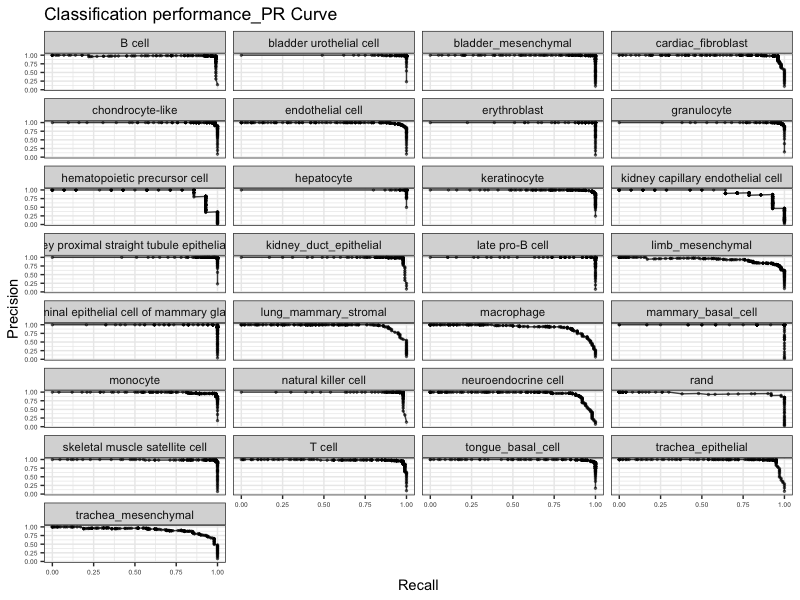
看一下全部细胞准确度和召回率的结果:
tm_heldoutassessment = assess_comm(ct_scores = classRes_val_all, stTrain = stTrain, stQuery = stTest, dLevelSID = "cell", classTrain = "newAnn", classQuery = "newAnn", nRand = 50)
plot_PRs(tm_heldoutassessment)
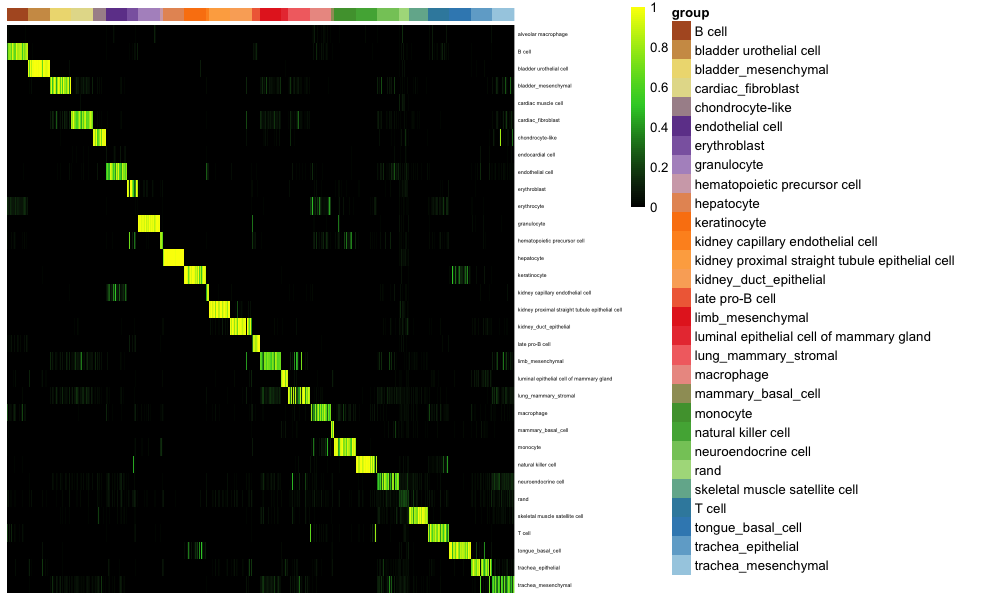
分类热图评分:
#Create a name vector label used later in classification heatmap where the values are cell types/ clusters and names are the sample names
nrand = 50
sla = as.vector(stTest$newAnn)
names(sla) = as.vector(stTest$cell)
slaRand = rep("rand", nrand)
names(slaRand) = paste("rand_", 1:nrand, sep='')
sla = append(sla, slaRand) #include in the random cells profile created
sc_hmClass(classMat = classRes_val_all,grps = sla, max=300, isBig=TRUE)
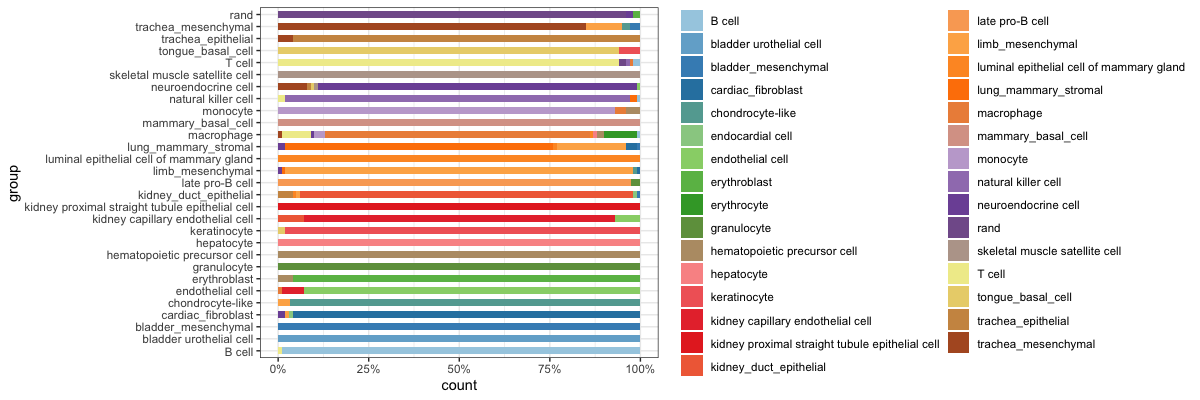
比例堆积图,这里如果分类效果好,细胞清晰的话理想状态是一行一个颜色,即每一组都是一种细胞类型
plot_attr(classRes=classRes_val_all, sampTab=stTest, nrand=nrand, dLevel="newAnn", sid="cell")
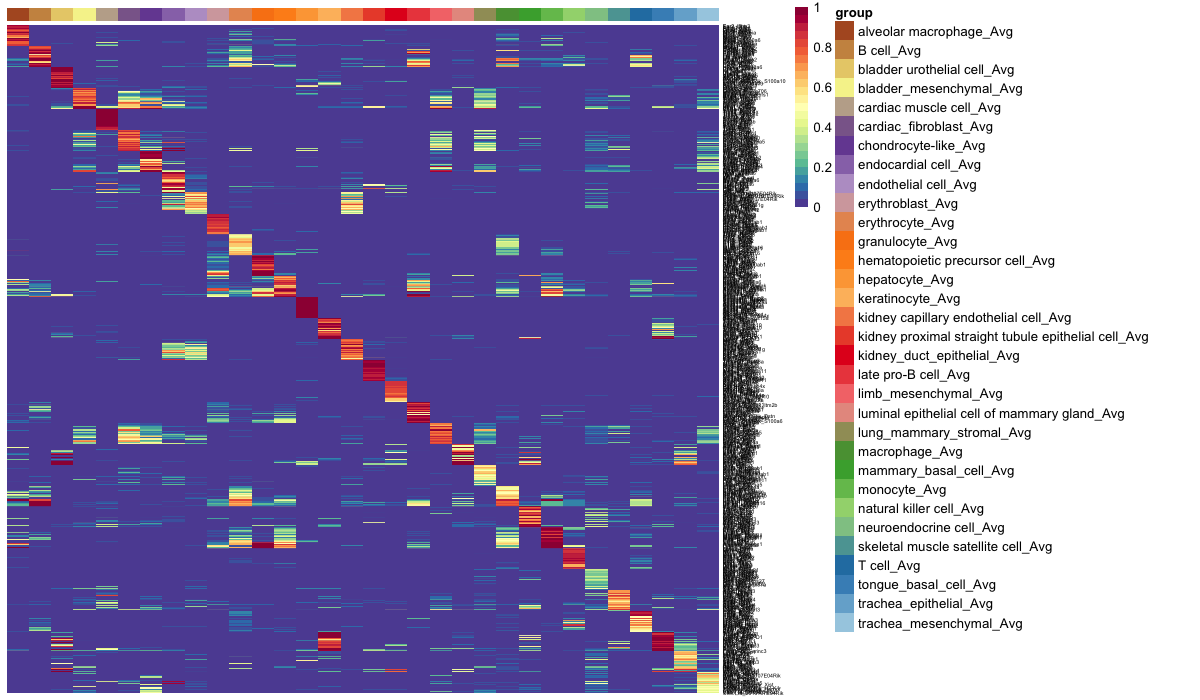
可视化训练数据的平均顶级对基因表达:
gpTab = compareGenePairs(query_exp = expTest, training_exp = expTrain, training_st = stTrain, classCol = "newAnn", sampleCol = "cell", RF_classifier = class_info$cnProc$classifier, numPairs = 20, trainingOnly= TRUE)
train = findAvgLabel(gpTab = gpTab, stTrain = stTrain, dLevel = "newAnn")
hm_gpa_sel(gpTab, genes = class_info$cnProc$xpairs, grps = train, maxPerGrp = 50)
查询数据及可视化:
expPark = utils_loadObject("expMatrix_Park_MouseKidney_Oct_12_2018.rda")
nqRand = 50
system.time(crParkall<-scn_predict(class_info[['cnProc']], expPark, nrand=nqRand))
# user system elapsed
# 89.633 5.010 95.041
sgrp = as.vector(stPark$description1)
names(sgrp) = as.vector(stPark$sample_name)
grpRand =rep("rand", nqRand)
names(grpRand) = paste("rand_", 1:nqRand, sep='')
sgrp = append(sgrp, grpRand)
# heatmap classification result
sc_hmClass(crParkall, sgrp, max=5000, isBig=TRUE, cCol=F, font=8)

分类注释分配:
# This classifies a cell with the catgory with the highest classification score or higher than a classification score threshold of your choosing.
# The annotation result can be found in a column named category in the query sample table.
stPark <- get_cate(classRes = crParkall, sampTab = stPark, dLevel = "description1", sid = "sample_name", nrand = nqRand)
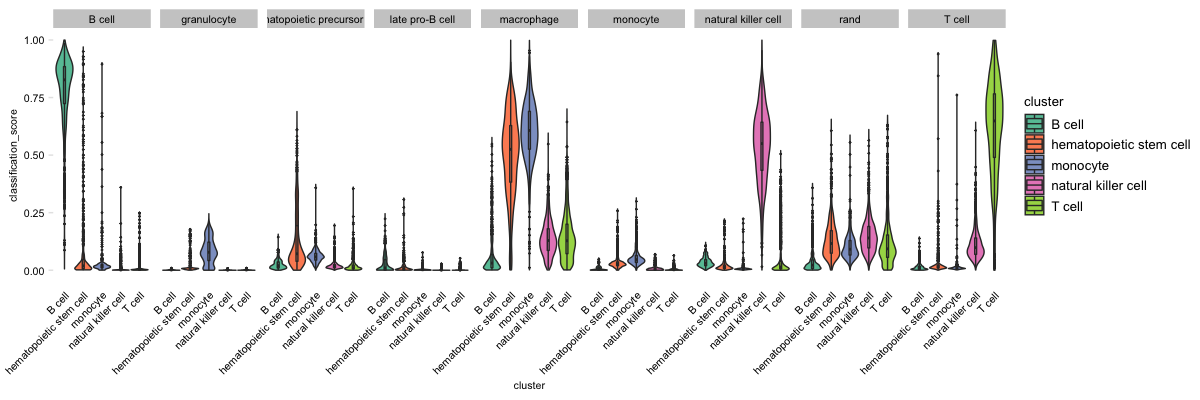
sc_violinClass(sampTab = stPark, classRes = crParkall, sid = "sample_name", dLevel = "description1", addRand = nqRand)

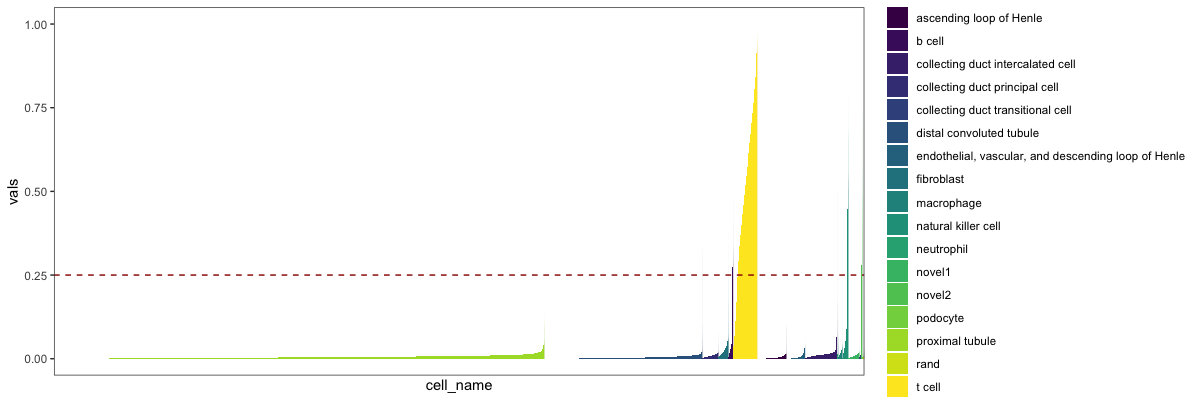
分类结果的天际线图,这个解释一下,可以看作是若干个细胞的密度图
library(viridis)
stKid2 = addRandToSampTab(crParkall, stPark, "description1", "sample_name")
skylineClass(crParkall, "T cell", stKid2, "description1",.25, "sample_name")
5.跨物种的评估
前面步骤是相似的,但是在加载直系同源物表时,将人类基因名称转换为小鼠直系同源物名称,并将分析限制为训练和查询数据之间共有的基因。
oTab = utils_loadObject("human_mouse_genes_Jul_24_2018.rda")
dim(oTab)
# [1] 16688 3
aa = csRenameOrth(expQuery, expTMraw, oTab)
expQueryOrth = aa[['expQuery']]
expTrainOrth = aa[['expTrain']]
各细胞类型横向的小提琴图:
sc_violinClass(sampTab = stQuery,classRes = crHS, sid = "sample_name", dLevel = "description", ncol = 12)
还有一个按分类划分的UMAP图:
system.time(umPrep_HS<-prep_umap_class(crHS, stQuery, nrand=nqRand, dLevel="description", sid="sample_name", topPC=5))
# user system elapsed
# 25.703 0.740 26.450
plot_umap(umPrep_HS)

6.小结
- 该工具的适用情况没有想象中的这么广,虽说是多物种特点,但一般研究都是分开做的。细胞分大类的时候用不上,细分小类亚型的时候又用不了。属于是附加项吧,但是需要自己构建特征数据集,再去验证评估自己的分类效果,还是有一点绕的。
- 因此小编认为还是bulk的适用更好一些,也不难怪作者对bulk的工具追更新了一版出来



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











