







高化
常用名(必记):
涤纶:聚对苯二甲酸乙二醇酯
腈纶:聚丙烯腈
维纶:聚乙烯醇(不是乙烯醇制备的,是醋酸乙烯酯的醇解,第九章提到)
聚氯乙烯:氯乙烯(只用温度控制聚合度,因为容易发生链转移)
尼龙-66:聚己二胺己二酸
天然橡胶:顺-1,4-聚异戊二烯
丁苯橡胶:丁二烯,苯乙烯
DPPH:1,1-二苯基-2-三硝基苯肼 用比色法定量,且是1:1消灭自由基
AIBN:偶氮二异丁腈
BPO:过氧化对苯二甲酸
每章需要思考的内容
- 反应机理
- 有无引发剂?引发剂分类?影响因素有什么?引发剂对什么能造成影响?
- 聚合度如何计算?什么因素能影响聚合度?
- 有什么特殊的反应和化合物需要记忆的?
缩聚和逐步聚合
连锁聚合
自由基聚合
机理
慢引发,快增长,速终止,有转移
引发剂先被引发,产生自由基,称为初级活性种;初级活性种与单体结合产生单体活性种;之后开始自由基聚合增长;有可能发生链转移反应;最后终止。
图形出现S型,说明存在自动加速现象
烯类单体对聚合机理的选择性
- 位阻效应
- 单取代,即使取代基再大也可以聚合
- 1,1-二取代,结构上不对称,一般可以形成高分子,如果取代集团太大,只能合成二聚体
- 1,2-二取代,结构对称,一般难合成,或只合成二聚体
- 特殊的一个物质就是氟元素,大小和氢差不多,因此位阻效应可以忽略,甚至还有电子效应
- 电子效应
-
无取代基
单体乙烯,难聚合,高温高压条件下自由基聚合,同时也会产生支链。 -
吸电子基
发生阴离子聚合,自由基聚合
如果吸电子效应太强,则只会发生阴离子聚合
特殊的卤原子,既有吸电效应,又有共轭效应,抵消了,因此只发生自由基聚合(除了氟离子) -
供电子基
发生阳离子聚合
烷基乙烯基醚,醚键的共轭效应比吸电效应强,因此属于供电基。 -
共轭效应
一般三种聚合方法都可进行
引发
引发剂
- 偶氮类
- 1.1 偶氮二异丁腈,偶氮二异庚腈
- 受温度影响大,加热可以产生气体,可以用作发泡剂
- 过氧化物
-
2.1 无机
过氧化氢,过硫酸钾或过硫酸铵 -
2.2 有机
- BPO 存在两段分解,受笼蔽效应的影响大,第二段分解产生苯自由基和二氧化碳。
- EHP 过氧化二碳酸二乙基己酯
- 氧化-还原体系
- 3.1 水溶性
氧化剂:过氧化氢,过硫酸盐,过氧化物
还原剂:有无机还原剂和有机还原剂
还原剂用量要比氧化剂的少
四价铈盐+醇酮胺,有效的发生单体聚合和接枝共聚
- 3.2 油溶性
常用:BPO/N,N-二甲基苯胺
氧化剂:过氧化二烷基,过氧化二酰,氢过氧化物
还原剂:有机金属化合物(三乙基铝,三乙基硼),硫醇(SH),
其他的引发方式:热引发,光引发,辐射引发,等离子体引发,微波引发
引发效率
有两种影响因素:
- 诱导分解
自由基向引发剂发生转移 - 笼蔽效应
引发剂处于单体或溶剂的笼子里,自由基寿命短,在此期间不发生反应就会分解,失去活性。
聚合速率的影响因素
这部分涉及到很多的重要的公式
链引发:
R
i
=
2
f
k
d
[
I
]
f
是
引
发
剂
效
率
R_i=2fk_d[I]\quad f是引发剂效率
Ri=2fkd[I]f是引发剂效率
为什么乘2? 因为一个引发剂产生两个自由基
链增长:
R
p
=
k
p
[
M
⋅
]
[
M
]
R_p=k_p[M·][M]
Rp=kp[M⋅][M]
链终止:
R
t
=
2
k
t
[
M
⋅
]
2
R_t=2k_t[M·]^2
Rt=2kt[M⋅]2
稳态:
R
i
=
R
t
R_i=R_t
Ri=Rt,
[
M
⋅
]
=
f
k
d
k
t
[
I
]
[M·]=\sqrt{\frac{fk_d}{k_t}[I]}
[M⋅]=ktfkd[I]
引发和终止这两部分的速率都非常得小,因此链增长为反应速率,但是自由基的浓度不好测定,要考虑如何在式中消去[M·]。这里要反复套用公式
[
M
⋅
]
=
f
k
d
k
t
[
I
]
[M·]=\sqrt{\frac{fk_d}{k_t}[I]}
[M⋅]=ktfkd[I]
R
p
=
k
p
[
M
]
∗
[
M
⋅
]
=
k
p
f
k
d
k
t
[
M
]
[
I
]
1
2
\begin{aligned} R_p&=k_p[M]*[M·] \\ &=k_p\sqrt{\frac{fkd}{kt}}[M][I]^\frac{1}{2} \end{aligned}
Rp=kp[M]∗[M⋅]=kpktfkd[M][I]21
- 温度(联系到阿伦尼乌兹公式)
k ′ = k p k d k t k'=k_p\frac{k_d}{k_t} \quad k′=kpktkd k ′ = A e − E / R T k'=Ae^{-E/RT}\quad k′=Ae−E/RT E = ( E p − E t 2 ) + E d 2 E=(E_p-\frac{E_t}{2})+\frac{E_d}{2} E=(Ep−2Et)+2Ed
E为正值,当温度升高,k’增大,速率增大 - 引发剂
转化率-时间图形中会出现:诱导期,初期,中期,后期。
其中最明显的是诱导期,是因为有阻聚剂的存在。初期匀速增长。中期自动加速。
自动加速现象
由于体系的粘度增大,自由基被包埋不能进行双基终止而不失活,活性仍然保持,速率不减反升
聚合度的影响因素
动力学链长和聚合度
动力学链长:一个活性种从链引发到链终止所消耗的单体分子数。
ν
=
R
p
R
i
=
R
p
R
t
=
k
p
2
(
f
k
d
k
t
)
1
2
[
M
]
[
I
]
1
2
\nu=\frac{R_p}{R_i}=\frac{R_p}{R_t}=\frac{k_p}{2(fk_dk_t)^{\frac{1}{2}}}\frac{[M]}{[I]^{\frac{1}{2}}}
ν=RiRp=RtRp=2(fkdkt)21kp[I]21[M]
聚合度:
无链转移:
X
n
ˉ
=
C
2
−
D
ν
\bar{X_n}=\frac{\frac{C}{2}-D}{\nu}\quad
Xnˉ=ν2C−D C表示偶合终止的百分比,D表示歧化终止的百分比
有链转移:
1
X
n
ˉ
=
C
2
+
D
ν
R
i
R
p
+
C
M
+
C
I
[
I
]
[
M
]
+
C
S
[
S
]
[
M
]
\frac{1}{\bar{X_n}}=\frac{\frac{C}{2}+D}{\nu}\frac{Ri}{R_p}+C_M+C_I\frac{[I]}{[M]}+C_S\frac{[S]}{[M]}
Xnˉ1=ν2C+DRpRi+CM+CI[M][I]+CS[M][S]
温度升高,聚合速率升高,但聚合度反而下降。
在聚合反应中,链增长反应速度越快,反应所得的聚合物分子量越大,但是聚合反应中,在温度升高的同时,链转移反应活化能的能量也随之增大,从而使链转移速度加快,所以反应温度高导致聚合物分子量下降。
阻聚剂:
-
加成型:苯醌,硝基化合物,氧,硫
苯醌最重要 -
链转移型:1,1-二苯基-2-三硝基苯肼(DPPH),芳胺,酚类
重要的是DPPH -
电荷转移型:变价金属的氯化物
推导自由基聚合动力学方程时,作了哪些基本假定?
在不考虑链转移反应的前提下,作了三个基本假定:
- 等活性假定,即链自由基的活性与链长无关;
- 稳态假定,即在反应中自由基的浓度保持不变;
- 聚合度很大假定。
一个重要的阻聚机理
烯丙基的自阻聚效应:
自由基p电子与π电子共轭,因共振而稳定,相互终止或与其他链自由基终止,类似于阻聚。
自由基共聚合
命名
无规共聚物:主单体-co-次单体
嵌段共聚物:先加入-b-后加入
交替共聚物:-alt-
接枝共聚物:主链-gra-支链
意义
用于改进大分子结构性能,增加品种,扩大应用范围。
五个假定:
- 分子量足够大
- 等活性理论
- 稳态
- 无前末端效应
- 不可逆聚合
基本方程
共聚物与单体瞬时组成:
d
[
M
1
]
d
[
M
2
]
=
M
1
M
2
⋅
r
1
M
1
+
M
2
r
2
M
2
+
M
1
\frac{d[M_1]}{d[M_2]}=\frac{M_1}{M_2}·\frac{r_1M_1+M_2}{r_2M_2+M_1}
d[M2]d[M1]=M2M1⋅r2M2+M1r1M1+M2
竞聚率r:
r
1
=
k
11
k
12
,
r
2
=
k
22
k
21
r_{1}=\frac{k_{11}}{k_{12}},r_2=\frac{k_{22}}{k_{21}}
r1=k12k11,r2=k21k22
f1(M1所占单体百分比):
f
1
=
[
M
1
]
[
M
1
]
+
[
M
2
]
f_1=\frac{[M_1]}{[M_1]+[M_2]}
f1=[M1]+[M2][M1]
F1(M1所占大分子百分比):
F
1
=
d
[
M
1
]
d
[
M
1
]
+
d
[
M
2
]
=
r
1
f
1
2
+
f
1
f
2
r
1
f
1
2
+
2
f
1
f
2
+
r
2
f
2
2
F_1=\frac{d[M1]}{d[M_1]+d[M_2]}=\frac{r_1f_1^2+f_1f_2}{r_1f_1^2+2f_1f_2+r_2f_2^2}
F1=d[M1]+d[M2]d[M1]=r1f12+2f1f2+r2f22r1f12+f1f2
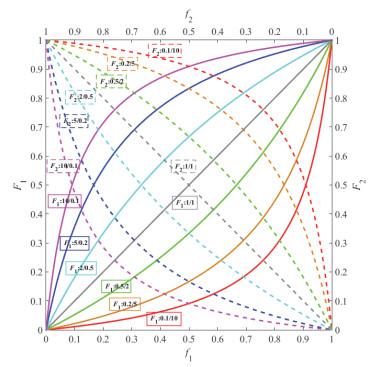
共聚曲线
r
1
r
2
=
1
r_1r_2=1
r1r2=1理想共聚:r1=r2时为对角线,
F
1
=
r
1
f
1
r
1
f
1
+
f
2
F_1=\frac{r_1f_1}{r_1f_1+f_2}
F1=r1f1+f2r1f1
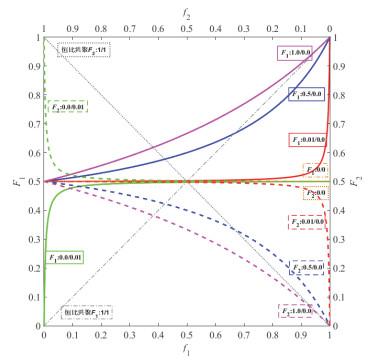
r
1
=
r
2
=
0
r_1=r_2=0
r1=r2=0交替共聚:
在0.5处的一条水平线
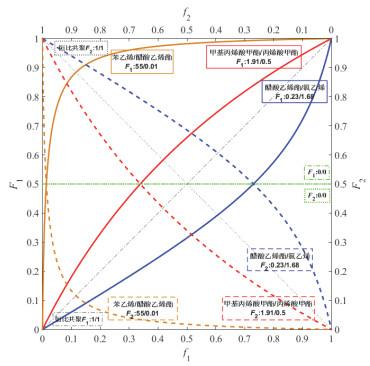
r
1
r
2
<
1
,
r
1
>
1
,
r
2
<
1
r_1r_2<1,r_1>1,r_2<1
r1r2<1,r1>1,r2<1非理想共聚:
在理想恒比共聚对角线以上部分,r1>1,
以下的部分,r2>1。
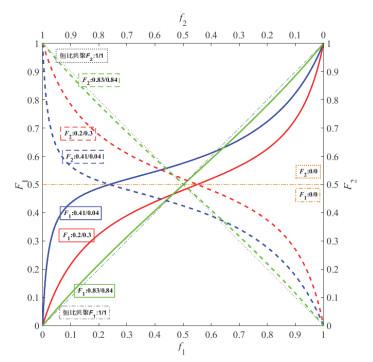
r
1
r
2
<
1
,
r
1
<
1
,
r
2
<
1
r_1r_2<1,r_1<1,r_2<1
r1r2<1,r1<1,r2<1非理想共聚:反S型,有恒比点
恒比点处:
[
M
1
]
/
[
M
2
]
=
d
[
M
1
]
/
d
[
M
2
]
[M_1]/[M_2]=d[M_1]/d[M_2]
[M1]/[M2]=d[M1]/d[M2],则:
r
1
[
M
1
]
+
[
M
2
]
=
r
2
[
M
2
]
+
[
M
1
]
r_1[M_1]+[M_2]=r_2[M_2]+[M_1]
r1[M1]+[M2]=r2[M2]+[M1]
那么
M
1
M
2
=
r
2
−
1
r
1
−
1
\frac{M_1}{M_2}=\frac{r_2-1}{r_1-1}
M2M1=r1−1r2−1
F
1
=
1
−
r
2
2
−
r
1
−
r
2
F_1=\frac{1-r_2}{2-r_1-r_2}
F1=2−r1−r21−r2
r
1
r
2
>
1
r_1r_2>1
r1r2>1 “嵌段”共聚:
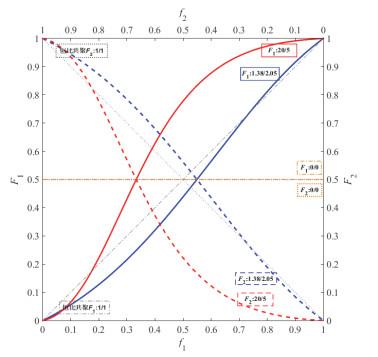
组成与转化率的关系:
定性描述:看图像中在理想恒比共聚对角线以上部分,f1向左移动(消耗M1速度快,f1应该减小),M1比M2活泼。
控制聚合物平均组成的方法:
- 控制转化率的一次投料法
- 补加活泼单体法
嵌段序列
生成一个x个M1的序列的概率: ( p M 1 ) = p 11 x − 1 p 12 (p_{M_1})=p_{11}^{x-1}p_{12} (pM1)=p11x−1p12
前末端效应
带有位阻或者极性基团的烯类单体进行自由基聚合时,前末端单元将对自由基活性产生影响
- 位阻效应
- 电子效应
特殊的反应物质:苯乙烯-丁烯二腈
Q-e
有一定的误差:因为将单体和自由基的e看成相等的了
Q:共轭效应度量,Q越大越容易变成自由基
e:极性度量
e值相差越大,越容易发生交替共聚
Q值相差越大,越难共聚
竞聚率
影响因素
温度
温度升高,向理想共聚物方向进行
压力
和温度类似,压力越大,向理想共聚物方向进行
溶剂
主要影响离子聚合,松紧对的影响,离子活性越高,定向能力越弱
其他因素
酸碱性,离子化程度
活性
单体活性:看
1
r
1
\frac{1}{r_1}
r11,越大,单体活性越高
自由基活性:看
k
12
=
k
11
r
1
k_{12}=\frac{k_{11}}{r_1}
k12=r1k11
一般而言,单体活性越高,自由基活性越小
离子聚合
这两种聚合方式都无凝胶效应,因为无法双基终止。
阴离子聚合
聚合单体
吸电子基,有π-π共轭
引发剂(Lewis碱,碱金属)
- 碱金属(电子转移)
1.1 直接转移
2.2 间接转移:苯乙烯-钠-萘-四氢呋喃
这个反应有个特殊的颜色反应:钠-萘-THF呈绿色,加入苯乙烯后呈现红色,反应完成后无色。双向引发,n=2 - 碱金属有机化合物
2.1 金属烷基化合物:丁基锂,格式试剂,催化效率高,单向引发n=1
2.2 碱金属氨基化合物:用得少,氨基钾
2.3 金属烷氧基化合物:
反应机理
快引发,快增长,不终止,不转移
活性聚合:反应不终止
原因:①末端都是阴离子,不能进行双基终止 ②反离子以金属离子为主,不能夺取H+ ③活性链中的H-离子难以被夺取,需要很高的能量
应用:①嵌段聚合物 ②合成分子量均一的聚合物 ③合成遥爪聚合物
判断是活性聚合的特征:
- 大分子具有活性末端,有再引发单体的能力
- 聚合度与单体浓度/起始引发剂浓度比值成正比
- 聚合物分子量随转化率线性增加
- 所有大分子同时增长,增长链数不变,聚合物分子量窄
如何使阴离子聚合终止?
特殊的链终止和链转移:可能自终止(端基异构化,形成不活泼的烯丙基型端基阴离子);可能与容器壁上杂质转移终止(向氨,甲苯,极性单体链转移);聚合末期要人为加入终止剂(含氧杂质:氧,水,二氧化碳)。
pKa比单体小的化合物均可使阴离子聚合终止。
Ka越小,pKa值越大,碱性越强,亲电能力越小。越难解离出H+。
用pKa来指导嵌段共聚物的合成。
先聚合pKa值大的。因为pKa大的可以引发小的,反之不成立。
动力学
反应速率: R p = − d M d t = k p [ C ] [ M ] l n [ M 0 ] [ M ] = k p [ C ] t R_p=-\frac{dM}{dt}=k_p[C][M]\\ln\frac{[M_0]}{[M]}=k_p[C]t Rp=−dtdM=kp[C][M]ln[M][M0]=kp[C]t
影响因素
- 溶剂:溶剂效应影响松紧对,越松聚合速率越快,但是定向能力越差
- 反离子:弱极性溶剂中,离子半径越大,溶剂化作用越明显;极性溶剂中,原子半径小的溶剂化作用更显著。
- 温度:解离平衡常数K随温度增大而减小。
阳离子聚合
反应单体
- α-烯烃,异丁烯:只有异丁烯可以反应,其他要高温高压下配位聚合
- 烷基乙烯基醚:存在p-π共轭,显示出给电子效应。
- 共轭烯烃
引发剂(Lewis酸,质子酸)
- 质子酸:
- Lewis酸:需要添加共引发剂做阳离子源
2.1 质子供体:
2.2 碳阳离子供体:RX,RCOX等卤代烷
聚合机理
快引发,快增长,难终止,易转移
转移:动力学链不终止,新的离子会继续反应
终止:①自发终止(链转移) ②与反离子加成 ③与反离子部分结合终止
影响因素
- 溶剂:溶剂效应影响松紧对,越松聚合速率越快,但是定向能力越差
- 反离子:亲核性过强,链终止;反离子体积大,松对,聚合速率大
- 温度:解离平衡常数K随温度增大而减小。温度低还能减弱链转移反应。
配位聚合
聚合方法

溶液聚合
单体和引发剂溶于适当的溶剂中聚合
离子聚合引发剂不耐水,不得不采用溶液聚合和淤浆聚合
单体浓度低和链转移反应,聚合度和聚合速率有所降低。
本体聚合
单体中加有少量引发剂的聚合
多采用油性引发剂:BPO,AIBN…
关键问题在于排热
可采用:间歇法,连续法
优点:
- 产品纯净,尤其适用于制透明板材、型材
- 聚合设备相对简单,可连续生产。
缺点:粘度大,聚合热不易排除,不易扩散,产生凝胶效应,自动加速,反应难控制
书上例子:苯乙烯连续本体聚合;甲基丙烯酸甲酯的间歇本体聚合;氯乙烯间歇本体聚合;乙烯高压本体聚合(高温高压:低密度聚乙烯)
这些聚合方式一般都分成两段聚合:①预聚:保持低转化率 ②聚合:转化率和粘度都较高,在特殊反应容器内进行
悬浮聚合
单体以液滴状悬浮在水中的聚合
基本组分:单体,引发剂,水,悬浮剂(能将油溶性单体悬浮在水中的物质)

悬浮剂:能在粒子表面形成保护层,降低表面张力,有利于液滴分散
(1)油溶性有机高分子(形成保护膜)
(2)不溶于水的无机粉末(机械隔离)
有时还要添加少量表面活性剂
多用的是油溶性引发剂,机理也与本体聚合相同
悬浮聚合初始体系是非均相,但是液滴小单元则是均相的
优点:
- 体系粘度低,传热和温度容易控制,分子量及分布稳定
- 产品分子量比溶液聚合的高,杂质比乳液聚合的少
- 后处理工序比乳液聚合和溶液聚合简单
缺点:
- 产物中带有少量分散剂残留物
- 难以实现连续化
书上例子:氯乙烯悬浮聚合;苯乙烯悬浮聚合;微悬浮聚合
影响颗粒形态的因素:

乳液聚合(很重要)
单体在水分中衡山城乳液状的聚合
特点:聚合速率和聚合度可以同时增加
优点:
- 水作为分散介质,散热容易
- 可以在低温下聚合
- Rp快,产物分子量高
缺点:
- 得到固体聚合物后处理麻烦,成本较高
- 难以除尽乳化剂及残留物
组成成分:单体,引发剂,水,乳化剂
- 乳化剂作用:①降低表面张力 ②在胶粒或液滴表面形成保护层,防止凝聚 ③形成胶束,使单体增溶
临界胶束浓度CMC:乳化剂开始形成胶束的浓度

- 乳化剂分类:
- 阴离子乳化剂(传统):极性基团是阴离子。
- 阳离子乳化剂:活性部分为阳离子
- 两性乳化剂
- 非离子乳化剂
脂肪酸纳,十二烷基硫酸钠
C
12
H
25
S
0
4
N
a
C_{12}H_{25}S0_4Na
C12H25S04Na,烷基磺酸钠,烷基芳基磺酸钠
用亲水亲油平衡值(HLB)表示亲水性大小。HLB越大越亲水。
常规乳液聚合乳化剂一般属于水包油型(O/W),在8~18范围内
乳液聚合三阶段:
- 成核期/增速期
成核机理:
- 胶束成核
-
胶粒数恒定期/恒速期
从增溶胶束消失开始,体系中只有胶粒和液滴 -
降速期
体系中已经无单体液滴,只剩下胶粒一种粒子



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









