






一、安装
VASPKIT下载:https://sourceforge.net/projects/vaspkit/files/Binaries/
(忘了从哪个版本开始,vaspkit结构转换部分被单独拿出来做了一个新的软件atomkit
ATOMKIT下载:https://sourceforge.net/projects/atomkit/files/Binaries/)
以vaspkit安装为例子,所有linux下的安装在集群上的软件包如下(Installation — VASPKIT 1.5 documentation):
第一步,如果压缩包软件以.gz结尾,需要先解压缩:$
tar -zxvf vaspkit.1.00.tar.gz第二步,配置环境,让你可以直接输入指令就可以使用:
cd vaspkit.1.00
cp -f how_to_set_environment_variable ~/.vaspkit此外,在这一步你可以设置vaspkit的一些默认参数,使用 vi ~/.vaspkit 可以修改一些vaspkit的设置
然后将vaspkit的绝对路径加入到环境变量当中(其中vaspkit.1.00需要替换为你安装的软件):
echo 'export PATH=/home/vaspkit.1.00/bin/:$PATH' >> ~/.bashrc
source ~/.bashrc你可以安装一些python的依赖包,例如官网上给出的。它们根据你的喜好和使用习惯,我跟习惯导出数据然后用origin这类软件做图。ATOMKIT同理。
二、使用
我们除了输入VASPKIT之后通过数字选择之外,还有几种选择方法输出我们所需要的指令(Tutorials — VASPKIT 1.5 documentation):
vaspkit -task 102 -kpr 0.04 #最为常用
echo -e "102\n2\n0.04\n"| vaspkit #更适合放在脚本里使用
(echo 102; echo 2; echo 0.04)|vaspkit三、翻译
下面以我个人的VASPKIT版本进行中文翻译
其中下面小标题带有(常用)标志的是,无论你是做什么方向,它们都应该掌握;如果是带*,那么你大概率会用到这些功能。
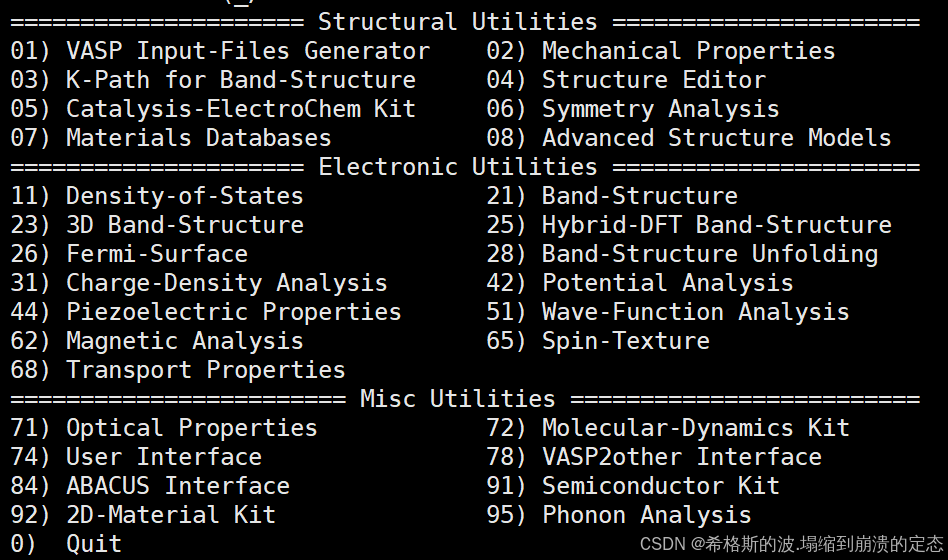
(一) Structural Utilities 结构工具(预处理)
这部分可以看作你要对计算数据的预处理,即需要处理之后才能计算,分为以下几类:
- VASP Input-Files Generator VASP 输入文件生成器
- Mechanical Properties 力学性质
- K-Path for Band-Structure 能带结构的 K 路径
- Structure Editor 结构编辑器
- Catalysis-ElectroChem Kit 催化-电化学工具包
- Symmetry Analysis 对称性分析
- Materials Databases 材料数据库
- Advanced Structure Models 高级结构模型
1. *VASP Input-Files Generator VASP 输入文件生成器(最常用)
其中包含了所有基础输入文件生成,即基础四文件(INCAR/KPOINTS/POSCAR格式转换/POTCAR):
101. Customize INCAR File 自定义 INCAR 文件
102. Generate KPOINTS File for SCF Calculation 为 SCF 计算生成 KPOINTS 文件
103. Generate POTCAR File with Default Setting 使用默认设置生成 POTCAR 文件
104. Generate POTCAR File with User Specified Potential 根据用户指定的势生成 POTCAR 文件
105. Generate POSCAR File from cif (no fractional occupations) 从 CIF 文件生成 POSCAR 文件(无分数占据)
106. Generate POSCAR File from Material Studio xsd (retain fixes) 从 Material Studio XSD 文件生成 POSCAR 文件(保留修正)
107. Reformat POSCAR File in Specified Order of Elements 按指定元素顺序重新格式化 POSCAR 文件
108. Successive Procedure to Generate VASP Files and Check 按步骤生成 VASP 文件并检查
109. Submit Job Queue 提交作业队列
其中,如果你使用vaspkit -task 102会生成默认精度为0.04 k-mesh的KPOINTS文件,并且会顺带检查补充四文件是否缺失然后自动补充(这个补充的文件是源于你VASPKIT的默认设置,即vi ~/.vaspkit 中你所设定的);此外vaspkit -task 103是你的标准赝势,一般我们默认使用此赝势。且一般情况下我们标准赝势是用的PBE泛函下的赝势。当然如果你需要使用超软/硬赝势对于某些特殊的体系,那么你可以使用vaspkit -task 104随意组合你的赝势。此外四文件里赝势文件是无法被覆盖的,这一点也需要注意。
重点说一下INCAR生成工具vaspkit -task 101,这个东西是很好用的,因为它里面包含了不同计算项目下的INCAR,且由于参与vaspkit软件编译的都是大佬,所以它生成的INCAR参数可以看作是权威的(比网上和不知道什么途径得来的好太多了):
计算选项
ST) Static-Calculation 静态计算
MG) Magnetic Properties 磁性性质
D3) DFT-D3 no-damping Correction DFT-D3 无阻尼校正
PU) DFT+U Calculation DFT+U 计算
GW) GW0 Calculation GW0 计算
DC) Elastic Constant 弹性常数
BD) Bader Charge Analysis Bader 电荷分析
EC) Static Dielectric Constant 静态介电常数
PH) Phonon-Calculation 声子计算
NE) Nudged Elastic Band (NEB) 微扰弹性带(NEB)
FQ) Frequence Calculation 频率计算
MT) Meta-GGA Calculation Meta-GGA 计算
SR) Standard Relaxation 标准弛豫
SO) Spin-Orbit Coupling 自旋轨道耦合
H6) HSE06 Calculation HSE06 计算
MD) Molecular Dynamics 分子动力学
BS) BSE Calculation BSE 计算
EL) ELF Calculation 电子局域函数计算
OP) Optical Properties 光学性质
PC) Decomposed Charge Density 分解电荷密度
PY) Phonon with Phonopy 使用 Phonopy 的声子计算
DM) The Dimer Method 二聚体方法
LR) Lattice Relaxation 晶格弛豫
PZ) Piezoelectric Calculation 压电计算
其中只输入vaspkit -task 101,那么默认为第一个选项(ST),它包含了大部分vaspkit结构优化,自洽,以及常用非自洽计算参数。其余参数根据你所需要即可。其中它生成的文件是只包含你关于这部分计算的参数,例如如果你想做一个加入HSE06杂化泛函的能带计算,你需要将PBE计算的INCAR + vaspkit生成HSE06的INCAR。这部分指令是cat pbe-INCAR >> vaspkit-INCAR。因此VASPKIT在这部分生成的INCAR全部默认文件名为“INCAR”,会覆盖你当前文件中的INCAR。此外,PY是DFPT(微扰法)方法的声子谱INCAR,PH是FD(有限位移法)方法的声子谱INCAR。
2. *Mechanical Properties 力学性质
这一部分涉及弹性常数的计算,通常用来判断结构存在与否:
200) Elastic-Constants Using Stress-Strain Method 使用应力-应变法计算弹性常数
201) Elastic-Constants Using Energy-Strain Method 使用能量-应变法计算弹性常数
202) Mechanical Properties from ELASTIC_TENSOR(_2D).in file 从 ELASTIC_TENSOR(_2D).in 文件计算力学性质
203) Elastic-Constants and Mechanical Properties from OUTCAR file 从 OUTCAR 文件计算弹性常数和力学性质
204) Spatial-Dependent Mechanics from ELASTIC_TENSOR(_2D).in file 从 ELASTIC_TENSOR(_2D).in 文件计算空间相关的力学性质
205) Equation-of-State Fitting 状态方程拟合
3. *K-Path for Band-Structure 能带结构的 K 路径(常用)
301) 1D Structure 一维结构
302) 2D Structure 二维结构
303) Bulk Structure 体相结构
304) K-Path for Wannier90 Code 为 Wannier90 代码生成 K 路径
305) K-Path for Phonopy Code 为 Phonopy 代码生成 K 路径
306) K-Path for CP2K Code 为 CP2K 代码生成 K 路径
309) Visualize K-Path in First Brillouin Zone 在第一布里渊区中可视化 K 路径
其中这部分用来得到能带的kpoints,二维就是302,块体就是303
如果需要计算声子谱,305可以生成声子谱的路径kpoints,309可以对布里渊区可视化。
其中如果需要计算杂化泛函的能带KPOINTS,那么明显不在这里,需要去杂化泛函部分寻找(vaspkit 的 25系列)
4. *Structure Editor 结构编辑器
这一部分是用来修改相关结构:
400) Redefine Lattice 重新定义晶格
401) Build Supercell 构建超胞
402) Fix Selected Atoms 固定选定原子
403) Move Selected Atoms 移动选定原子
404) Delete Selected Atoms 删除选定原子
405) Swap Axis of Lattice Vectors 交换晶格矢量的轴
406) Sort Atomic Coordinates in Specified Direction 按指定方向排序原子坐标
407) Rotate Lattice Vectors 旋转晶格矢量
408) Rotate Selected Atoms about Specified Rotation Axis 绕指定旋转轴旋转选定原子
409) Add Atom in Specified Position 在指定位置添加原子
410) Substitute Selected Atoms 替换选定原子
411) Copy Selected Atoms from Another Structure File 从其他结构文件复制选定原子
412) Apply Random-Displacements on Selected Atoms 对选定原子应用随机位移
413) Convert Between Fractional and Cartesian Coordinates 在分数坐标与笛卡尔坐标之间转换
414) Remove Spurious Lattice-Distortion after Optimization 优化后移除虚假晶格畸变
415) Apply Mirror Transformation in Specified Plane Mirror 在指定平面镜中应用镜像变换
419) Export to Other Structure Formats 导出为其他结构格式
5.Catalysis-ElectroChem Kit 催化-电化学工具包
我完全没用过:
501) Thermal Corrections for Adsorbate 吸附物的热修正
502) Thermal Corrections for Gas 气体的热修正
503) Band Center (experimental) 带心(实验)
504) Convert NEB-Path to PDB Format for Animation 将 NEB 路径转换为 PDB 格式以用于动画
505) Interpolate NEB Images Linearly 线性插值 NEB 图像
507) Imaginary Frequencies Correction 虚频修正
508) Bader2PQR (Shown in VMD by atomic charge) Bader2PQR(在 VMD 中按原子电荷显示)
509) Evaluate Half-Life Period for a First Order Reaction 评估一级反应的半衰期
6. *Symmetry Analysis 对称性分析(常用)
601) Find Symmetry of Crystal 查找晶体的对称性
602) Find Primitive Cell 查找原胞
603) Find Standard Conventional Cell 查找晶胞
604) Find Symmetrically Equivalent Atoms 查找对称等价的原子
608) Find Symmetry of Relaxed-Structure 查找弛豫结构的对称性
609) Find Symmetry of Molecule or Cluster 查找分子或簇的对称性
601是找对称性,它的精细度标准是你在vaspkit里根据SYMMERC设置的;602和603是找对应结构的原胞和晶胞,它们找到的结构会生成新的文件(一定要注意)。其余三个都是只找对称性。
7.Materials Databases 材料数据库
702) Computational 2D Monolayer Semiconductors 计算二维单层半导体
705) Computational 2D Semiconductor Heterostructures 计算二维半导体异质结构
这个是为了迎合当下高通量筛选,vaspkit自己做的一个数据库,张富盛师兄也是根据此内容完成的文章。其中VASPKIT相关材料信息数据库网址为:2D Semiconductor Database — Materials Database documentation
8.Advanced Structure Models 高级结构模型
太高级了,完全没用过:
800) Build Orthogonal Supercell 构建正交超胞
801) Build Vacuum Slab in Specified Direction 在指定方向构建真空层
802) Build Random Substitutional Alloy 构建随机置换合金
803) Build Surface by Specified Miller Indices 通过指定米勒指数构建表面
804) Build Heterostructure by Two Specified Slabs 通过两个指定薄膜构建异质结构
806) Build Quantum-Dot by Specified Radius 构建指定半径的量子点
807) Build NanoWire/2D Quantum-Dot by Specified Radius 构建指定半径的纳米线/二维量子点
808) Build Nanotube by Specified Orthogonal Slab 通过指定正交薄膜构建纳米管
其中还有一些pro的功能,估计需要氪金才能用。
(二) Electronic Utilities 电子工具(后处理)
这一部分可以常用来对已经计算完成的数据进行二次处理或者后处理,分为几类:
- Density-of-States 态密度
- Band-Structure 能带结构
- 3D Band-Structure 三维能带结构
- Hybrid-DFT Band-Structure 混合 DFT 能带结构
- Band-Structure Unfolding 能带展开
- Fermi-Surface 费米面
- Charge-Density Analysis 电荷密度分析
- Potential Analysis 势分析
- Piezoelectric Properties 压电性质
- Wave-Function Analysis 波函数分析
- Magnetic Analysis 磁性分析
- Spin-Texture 自旋
- Transport Properties 输运性质
1. *Density-of-States 态密度(常用)
用以描述原子的对于整体能带的贡献,类似于投影能带,可以与投影能带形成互补:
110) Inverse Participation Ratio 反参与比
111) Total Density-of-States 总态密度
112) Projected Density-of-States of Selected One Atom 选定单个原子的投影态密度
113) Projected Density-of-States of Each Element 各元素的投影态密度
114) Projected Density-of-States of Selected Atoms 选定原子的投影态密度
115) Projected Density-of-States of Selected Atoms and Orbitals 选定原子和轨道的投影态密度
116) Local Density-of-States of Each Element 各元素的局域态密度
117) Total Density-of-States from EIGENVAL File 从 EIGENVAL 文件提取的总态密度
118) Projected Density-of-States from EIGENVAL and PROCAR Files 从 EIGENVAL 和 PROCAR 文件提取的投影态密度
119) Projected Density-of-States of Specified K-Indexes 指定 K 指数的投影态密度
120) Projected Density-of-States of Specified Band-Indexes 指定能带指数的投影态密度
123) 3D Spatially-Resolved DOS in Specified Energy Range 在指定能量范围内的三维空间分辨态密度
124) 3D Spatially-Resolved Magnetic DOS in Specified Energy Range 在指定能量范围内的三维空间分辨磁态密度
125) 2D Plane-Averaged Spatially-Resolved DOS 平面平均二维空间分辨态密度
126) 2D Plane-Averaged Spatially-Resolved Magnetic DOS 平面平均二维空间分辨磁态密度
其实从名字上就能看出个大概。111给出总的态密度,112和114为原子投影,在分析异质结的时候可以作为层投影;113是给出各元素投影。其余按名字可以参考选取。
2. *Band-Structure 能带结构 (常用)
和态密度以及投影态密度类似,其实看名字就可以了解。
211) Band-Structure 能带结构
212) Projected Band-Structure of Only-One-Selected Atom 选定单个原子的投影能带结构
213) Projected Band-Structure of Each Element 各元素的投影能带结构
214) Projected Band-Structure of Selected Atoms 选定原子的投影能带结构
215) Projected Band-Structure by Element-Weights 按元素权重的投影能带结构
216) The Sum of Projected Band for Selected Atoms and Orbitals 选定原子和轨道的投影能带总和
这部分,如果你想要知道杂化泛函的能带结构那么需要去隔壁的“Hybrid-DFT Band-Structure 混合 DFT 能带结构”查询。
3. 3D Band-Structure 三维能带结构
这个其实不常用,因为我们通常计算2D材料
231) Generate KPOINTS File for 3D Band-Structure 为三维能带结构生成 KPOINTS 文件
232) 3D Band-Structure for 2D Material 二维材料的三维能带结构
233) 3D Band-Structure of HOMO & LUMO Bands [ISPIN=1 ONLY] HOMO 和 LUMO 带的三维能带结构 [仅限 ISPIN=1]
4. *Hybrid-DFT Band-Structure 杂化泛函 DFT 能带结构(常用)
如果你需要计算杂化泛函的能带结构,那么请看他。这里面有详细的指导。
250) Generate KPOINTS Including Irreducible Kmesh and Band Edges 生成包含不可约 K 网格和带边的 KPOINTS 文件
251) Generate KPOINTS for Hybrid Band-Structure 为混合能带结构生成 KPOINTS 文件
252) Band-Structure for Hybrid-DFT Calculation 混合 DFT 计算的能带结构
253) Projected Band-Structure for Selected One Atom 选定单个原子的投影能带结构
254) Projected Band-Structure for Each Element 各元素的投影能带结构
255) Projected Band-Structure for Selected Atoms 选定原子的投影能带结构
256) Projected Band-Structure by Element-Weights 按元素权重的投影能带结构
257) Sum of Projected Band for Selected Atoms and Orbitals 选定原子和轨道的投影能带总和
5. Band-Structure Unfolding 能带展开
这部分处理能带反折叠
281) Generate KPOINTS File for Band-Unfolding Calculation 为能带展开计算生成 KPOINTS 文件
282) Effective Band Structure (EBS) 有效能带结构 (EBS)
283) Orbital-Projected EBS for Selected Atoms 选定原子的轨道投影 EBS
284) Orbital-Projected EBS for Each Element 各元素的轨道投影 EBS
285) Sum of Orbital-Projected EBS for Selected Atoms & Orbitals 选定原子和轨道的轨道投影 EBS 总和
6. Fermi-Surface 费米面
完全没用过,或许你们会用到
261) Generate KPOINTS File for Fermi-Surface Calculation 为费米面计算生成 KPOINTS 文件
262) Fermi-Surface with XcrySDen format 以 XcrySDen 格式生成费米面
263) Fermi-Surface with FermiSurfer format 以 FermiSurfer 格式生成费米面
264) Projected Fermi-Surface with FermiSurfer format 以 FermiSurfer 格式生成投影费米面
265) Sum of Projected Fermi-Surface for Selected Atoms & Orbitals 选定原子和轨道的投影费米面总和
266) Fermi-Surface for 2D Materials 二维材料的费米面
267) Projected Fermi-Surface for 2D Materials 二维材料的投影费米面
7. *Charge-Density Analysis 电荷密度分析
1. Charge Options (电子密度选项): 这些选项与电子密度或相关性质的计算与分析有关:
310) Slice of Charge Density 提取特定平面上的电子密度切片。
311) Charge Density 提取整体电子密度分布。
312) Spin Density 提取自旋密度分布。
313) Spin-Up & Spin-Down Density 提取自旋向上和向下的密度分布。
314) Charge-Density Difference 计算电子密度差分,例如用于分析吸附体系的电子转移。
315) Linear-Average Charge Density 计算电子密度的线性平均值。
316) Planar-Average Charge Density 计算电子密度的平面平均值。
317) Macroscopic-Average Charge Density 计算电子密度的宏观平均值。
318) Charge Density Along Specified Path 沿特定路径计算电子密度分布。
2. Advanced Options (高级选项):这些选项与高级处理和数据格式转换有关:
320) Build Supercell of Charge Density by Transformation Matrix 通过变换矩阵构建超胞的电子密度。
325) Scanning Tunneling Microscope (STM) Simulation 模拟扫描隧道显微镜(STM)图像。
328) Export CHGCAR/PARCHG to XcrysDen .xsf format 将 CHGCAR 或 PARCHG 数据导出为 XcrysDen 使用的 .xsf 格式。
329) Export CHGCAR/PARCHG to Gaussian .cube format 将 CHGCAR 或 PARCHG 数据导出为 Gaussian 使用的 .cube 格式
这部分电荷密度我们常用到314,316. 其中314用来可视化差分电荷密度,316用来可视化电子密度的平面,比如我们用电荷密度方法计算介电材料的厚度就是通过316功能,也通常简称为‘电荷密度‘。
8. *Potential Analysis 势分析
420) Slice of Potential 势的切片(这个翻译感觉多少有点问题)
422) Linear-Average Potential 线性平均势
425) Potential Along Specified Path 沿指定路径的势
426) Planar-Average Potential 平面平均势
427) Macroscopic-Average Potential 宏观平均势
428) Export LOCPOT/ELFCAR to XcrysDen .xsf format 将 LOCPOT/ELFCAR 导出为 XcrysDen 的 .xsf 格式
429) Export LOCPOT/ELFCAR to Gaussian .cube format 将 LOCPOT/ELFCAR 导出为 Gaussian 的 .cube 格式
430) Build Supercell of Potential by Transformation Matrix 通过变换矩阵构建势的超胞
这部分我们常用426计算静电势,也从这里可以得到相应材料的真空能级。想使用这个功能,需要在INCAR中加入LVHAR这个参数。如果想得到异质结的真空能级,那么我们需要将异质结两侧的静电势取平均。
9. *Wave-Function Analysis 波函数分析
511) Wave-Function in Real-Space with CHGCAR format 实空间波函数 (CHGCAR 格式)
512) Wave-Function in Real-Space with Cube format 实空间波函数 (Cube 格式)
513) Wave-Function Parity (experimental) 波函数奇偶性 (实验)
515) Wave-Function Squared in Real-Space with CHGCAR format 实空间波函数平方 (CHGCAR 格式)
516) Wave-Function Squared in Real-Space with Cube format 实空间波函数平方 (Cube 格式
我们在这里使用511较多,可以直接生成选定k点和能带的波函数可视化(会生成一个.vasp文件)
10. Piezoelectric Properties 压电性质
442) Piezoelectric and Dielectric Constants from OUTCAR file 从 OUTCAR 文件提取压电和介电常数
443) Piezoelectric Properties 压电性质
11. Magnetic Analysis 磁性分析
621) Magnetic Anisotropy Energy 磁各向异性能量
629) Visualize Magnetic Moment Vectors 可视化磁矩矢量
我们很少计算带磁性材料,一般如果是金属或者是新型材料可能会用到
12. Spin-Texture 自旋
651) Generate KPOINTS File for Spin-Texture 为自旋纹理生成 KPOINTS 文件
652) Spin-Texture of Specified Band for 2D Material (SOC ONLY) 二维材料指定能带的自旋纹理 (仅限 SOC)
653) Spin-Texture of Specified Band for Bulk Material (SOC ONLY) 体材料指定能带的自旋纹理 (仅限 SOC)
这部分主要涉及到计算自旋轨道耦合,一般我们计算的材料涉及不到
13. Transport Properties 输运性质
681) Generate KPOINTS File for Transport Calculation 为输运计算生成 KPOINTS 文件
682) Transport Properties Based on Boltzmann Theory 基于玻尔兹曼理论的输运性质
输运性质我们也基本不用
(三) Misc Utilities杂项工具
- Optical Properties 光学性质
- Molecular-Dynamics Kit 分子动力学工具包
- User Interface 用户界面
- VASP2other Interface VASP 转换工具接口
- ABACUS Interface ABACUS 接口
- 2D-Material Kit 二维材料工具包
- Semiconductor Kit 半导体工具包
- Phonon Analysis 声子分析
1. Optical Properties 光学性质
我们算光学性质算的不多,当然回头会单独讲如何计算光学性质,会用到这部分
710) Linear Optical Spectrums for Two-Dimensional Semiconductors 二维半导体的线性光谱
711) Linear Optical Spectrums for Bulk Semiconductors 体半导体的线性光谱
713) Transition Dipole Moment from WAVECAR file 从 WAVECAR 文件提取跃迁偶极矩
714) Dipole Moment Elements from WAVEDER file 从 WAVEDER 文件提取偶极矩元素
716) Total Joint Density of States 总联合态密度
717) Partial Joint Density of States between Two Bands 两个能带之间的部分联合态密度
719) Spectroscopic Limited Maximum Efficiency 光谱限制的最大效率
2. Molecular-Dynamics Kit 分子动力学工具包
类似的,我们通常分子动力学做的比较简单,只是看材料的热稳定性,所以这部分内容基本上用不上
721) Mean Squared Displacement 均方位移
722) Mean Squared Displacement Using FFT (Recommend) 使用 FFT 计算均方位移(推荐)
723) Diffusion Coefficient & Ion Mobility from MSD.dat File 从 MSD.dat 文件提取扩散系数和离子迁移率
725) Pair Correlation Function from PCDAT File 从 PCDAT 文件提取对关联函数
726) Radial Distribution Function of Selected Elements 选定元素的径向分布函数
727) Velocity Autocorrelation Function 速度自相关函数
728) Vibrational DOS from Velocity Autocorrelation Function 从速度自相关函数计算振动态密度
730) Bond Length Distribution of Selected Elements 选定元素的键长分布
731) Bond Angle Distribution of Selected Elements 选定元素的键角分布
736) MD Trajectories of Selected Atoms with POSCAR Format 选定原子的分子动力学轨迹(POSCAR 格式)
737) MD Trajectories of Selected Atoms with PDB Format 选定原子的分子动力学轨迹(PDB 格式)
3. User Interface 用户界面
u1) Get Entropy for Adsorbate 计算吸附物的熵
u2) Bader2PQR Bader 电荷分析结果转换为 PQR 格式
4. VASP2other Interface VASP 转换工具接口 & ABACUS Interface ABACUS 接口
781) VASP2BoltzTraP Interface VASP 到 BoltzTraP 的接口
788) Normalized Trace of Interatomic Force Constant Tensors 原子间力常数张量的归一化迹
789) Sort Phonon Band Structure for Phonopy 为 Phonopy 排序声子能带结构
201) Elastic-Constants Using Energy-Strain Method 使用能量-应变法计算弹性常数
5. *2D-Material Kit 二维材料工具包
我们使用911获取材料的带隙,带边位置与能量
911) Band-Gap 带隙
912) Effective-Mass of Carrier 载流子的有效质量
913) Effective-Mass of HOMO & LUMO Bands [ISPIN=1 ONLY] HOMO 和 LUMO 带的有效质量 [仅限 ISPIN=1]
914) Angular-Dependent Effective-Mass and Fermi-Velocity 角度相关的有效质量和费米速度
917) Fermi Velocity of Carrier 载流子的费米速度
6. *Semiconductor Kit 半导体工具包
920、921移动原子在材料的位置很常用。当然 MS,修改POSCAR也可以做到。
920) Move Atomic-Layer to the bottom in z direction 将原子层移动到 z 方向的底部
921) Center Atomic-Layer in z direction 将原子层置于 z 方向的中心
922) Resize Vacuum Thickness 调整真空层厚度
923) Standardize 2D Crystal Cell 标准化二维晶胞
926) Stacking-dependent Potential Energy Surface 堆垛依赖的势能面
927) Band Edges Referenced to Vacuum Level for 2D Structure 参考真空能级的二维结构能带边缘
929) Summary for Relaxed 2D Structure 弛豫二维结构的总结
7. Phonon Analysis 声子分析
952) Phonon Dispersions From OUTCAR file 从 OUTCAR 文件提取声子色散
953) Projected Phonon Dispersion for Each Element 各元素的投影声子色散
954) Projected Phonon Dispersion for Selected Atoms 选定原子的投影声子色散
955) Projected Phonon Dispersion by Element-Weights 按元素权重的投影声子色散
(四) ATOMKIT分析
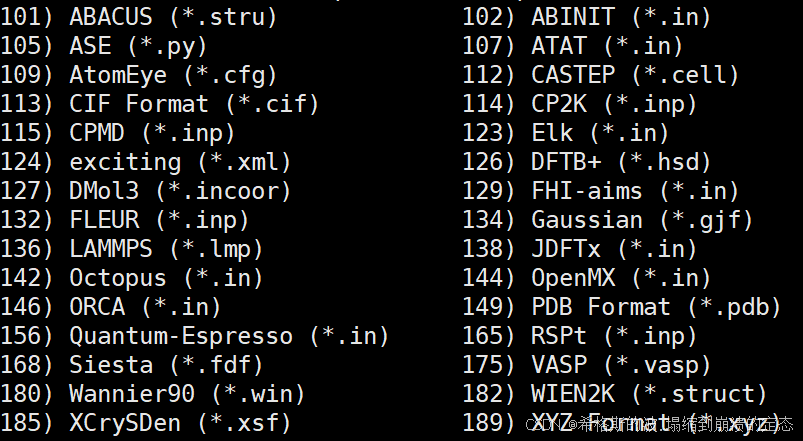
这个软件唯一和vaspkit区别的点在于,它的文件转换功能。即你可能同时需要使用多个软件进行运算,所以会需要将类似POSCAR转换为其他输入文件的格式。
和VASPKIT一样,调用指令 $ atomkit
-
ABACUS (.stru)
适用软件:ABACUS
中文翻译:ABACUS 结构文件
介绍:ABACUS 是一款基于从头计算的材料模拟软件,.stru文件用于定义晶体结构信息,包括原子类型、坐标和晶胞参数。 -
ABINIT (.in)
适用软件:ABINIT
中文翻译:ABINIT 输入文件
介绍:ABINIT 是一个多功能的第一性原理计算软件,.in文件包含计算所需的参数、初始条件和材料的相关信息。 -
ASE (.py)
适用软件:ASE (Atomic Simulation Environment)
中文翻译:原子模拟环境 Python 脚本
介绍:ASE 是一个用 Python 编写的开源工具,用于设置、执行和分析原子模拟,.py文件通常用于运行自定义模拟脚本。 -
ATAT (.in)
适用软件:ATAT
中文翻译:ATAT 输入文件
介绍:ATAT 是一个用于研究合金热力学和结构稳定性的工具,.in文件提供了用于计算的输入参数。 -
AtomEye (.cfg)
适用软件:AtomEye
中文翻译:AtomEye 配置文件
介绍:AtomEye 是一个快速的三维材料结构可视化工具,.cfg文件定义了原子类型、坐标和相关的结构参数。 -
CASTEP (.cell)
适用软件:CASTEP
中文翻译:CASTEP 结构文件
介绍:CASTEP 是一个基于第一性原理的材料模拟工具,.cell文件包含晶体结构和计算相关的几何信息。 -
CIF Format (.cif)
适用软件:多种晶体学软件(如 VESTA、Materials Studio)
中文翻译:晶体信息文件
介绍:CIF 文件是国际通用的晶体结构描述格式,记录了晶胞参数、对称性和原子位置等信息。 -
CP2K (.inp)
适用软件:CP2K
中文翻译:CP2K 输入文件
介绍:CP2K 是一款用于从头计算和分子动力学模拟的软件,.inp文件定义了模拟参数、材料结构和计算设置。 -
CPMD (.inp)
适用软件:CPMD
中文翻译:CPMD 输入文件
介绍:CPMD 专注于分子动力学和电子结构计算,.inp文件包含了所有的输入参数和初始设置。 -
Elk (.in)
适用软件:Elk
中文翻译:Elk 输入文件
介绍:Elk 是一个全对称第一性原理计算软件,.in文件用于定义计算所需的材料参数和方法。 -
DFTB+ (.hsd)
适用软件:DFTB+
中文翻译:DFTB+ 文件
介绍:DFTB+ 是基于紧束缚方法的量子力学计算工具,.hsd文件包含了模型参数和模拟设置。 -
exciting (.xml)
适用软件:exciting
中文翻译:exciting 输入文件
介绍:exciting 是基于全势 LAPW 方法的第一性原理计算工具,.xml文件用于设置材料模拟的参数。 -
FHI-aims (.in)
适用软件:FHI-aims
中文翻译:FHI-aims 输入文件
介绍:FHI-aims 是一个适用于材料和分子模拟的从头计算工具,.in文件用于定义原子结构和计算参数。 -
FLEUR (.inp)
适用软件:FLEUR
中文翻译:FLEUR 输入文件
介绍:FLEUR 是基于 LAPW 方法的电子结构计算软件,.inp文件用于设置材料和模拟参数。 -
Gaussian (.gjf)
适用软件:Gaussian
中文翻译:Gaussian 输入文件
介绍:Gaussian 是一种常用的量子化学计算工具,.gjf文件用于定义分子结构、计算方法和参数。 -
LAMMPS (.lmp)
适用软件:LAMMPS
中文翻译:LAMMPS 脚本文件
介绍:LAMMPS 是一个分子动力学模拟软件,.lmp文件包含模拟的初始条件、参数和计算设置。 -
JDFTx (.in)
适用软件:JDFTx
中文翻译:JDFTx 输入文件
介绍:JDFTx 是一个开放源代码的密度泛函理论计算工具,.in文件定义了计算参数和初始条件。 -
Octopus (.in)
适用软件:Octopus
中文翻译:Octopus 输入文件
介绍:Octopus 是一个适用于时间依赖性密度泛函理论的计算工具,.in文件用于定义计算的材料和方法。 -
OpenMX (.in)
适用软件:OpenMX
中文翻译:OpenMX 输入文件
介绍:OpenMX 是一个紧束缚线性组合原子轨道法的计算工具,.in文件定义了计算设置和初始条件。 -
PDB Format (.pdb)
适用软件:多种分子可视化软件(如 PyMOL、VMD)
中文翻译:蛋白质数据银行格式
介绍:PDB 文件是蛋白质和分子结构常用的标准格式,记录了原子的三维坐标和化学信息。 -
Quantum-Espresso (.in)
适用软件:Quantum ESPRESSO
中文翻译:Quantum ESPRESSO 输入文件
介绍:Quantum ESPRESSO 是一个电子结构计算软件,.in文件用于定义计算设置和晶体结构。 -
RSPt (.inp)
适用软件:RSPt
中文翻译:RSPt 输入文件
介绍:RSPt 是一种基于密度泛函理论的全势计算工具,.inp文件用于设置模拟参数。 -
Siesta (.fdf)
适用软件:Siesta
中文翻译:Siesta 输入文件
介绍:Siesta 是一个基于紧束缚方法的材料模拟软件,.fdf文件包含输入参数和结构定义。 -
VASP (.vasp)
适用软件:VASP
中文翻译:VASP 文件
介绍:VASP 是一个广泛使用的从头计算软件,.vasp文件定义了晶体结构信息。 -
Wannier90 (.win)
适用软件:Wannier90
中文翻译:Wannier90 输入文件
介绍:Wannier90 是用于构造最大局域化 Wannier 函数的工具,.win文件用于定义计算参数。 -
WIEN2K (.struct)
适用软件:WIEN2K
中文翻译:WIEN2K 结构文件
介绍:WIEN2K 是基于 LAPW 方法的材料模拟软件,.struct文件描述晶体结构信息。 -
XCrySDen (.xsf)
适用软件:XCrySDen
中文翻译:XCrySDen 文件
介绍:XCrySDen 是一个材料结构可视化工具,.xsf文件用于表示晶体结构和电子分布。 -
XYZ Format (.xyz)
适用软件:多种分子模拟软件(如 ASE、VMD)
中文翻译:XYZ 格式
介绍:XYZ 文件是描述分子结构的简单格式,记录了原子类型及其三维坐标


















 2586
2586

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









