







零基础入门转录组分析——数据处理(TCGA数据库)
目录
TCGA应该是肿瘤数据最权威的来源之一,但是从TCGA上下载数据集相对来说比较麻烦,因此出现了很多针对TCGA数据进行二次开发的衍生网站,XENA.UCSC就是很直观强大的一个在线网站,里面收录了众多数据库的数据集,其中就包括了TCGA数据集,并且是整合好的,可以直接用于分析。
并且XENA.UCSC这个网站不仅能下载数据,还能进行在线分析,例如:KM分析,表达量分析等,详细情况可以参考好用的TCGA分析工具:UCSC Xena,但是在这篇教程中仅介绍如何从UCSC上下载所需要的TCGA数据集,并且下载后在R中对数据集进一步处理成后续分析所要的形式。
本项目以非小细胞肺癌(non-small cell lung cancer,NSCLC)中的肺腺癌(lung adenocarcinoma,LUAD)作为展示
选用的数据库是TCGA。
物种:人类(Homo sapiens)
实验分组:疾病组,对照组。
我这里使用的R版本是4.2.2
要用到的R包:tidyverse, rtracklayer, dplyr
1. 数据集获取
首先进入XENA.UCSC官网(如下图所示),点击箭头指向的位置进一步运行Xena。

进一步点击DATA SETS进入Xena中存储数据集的页面
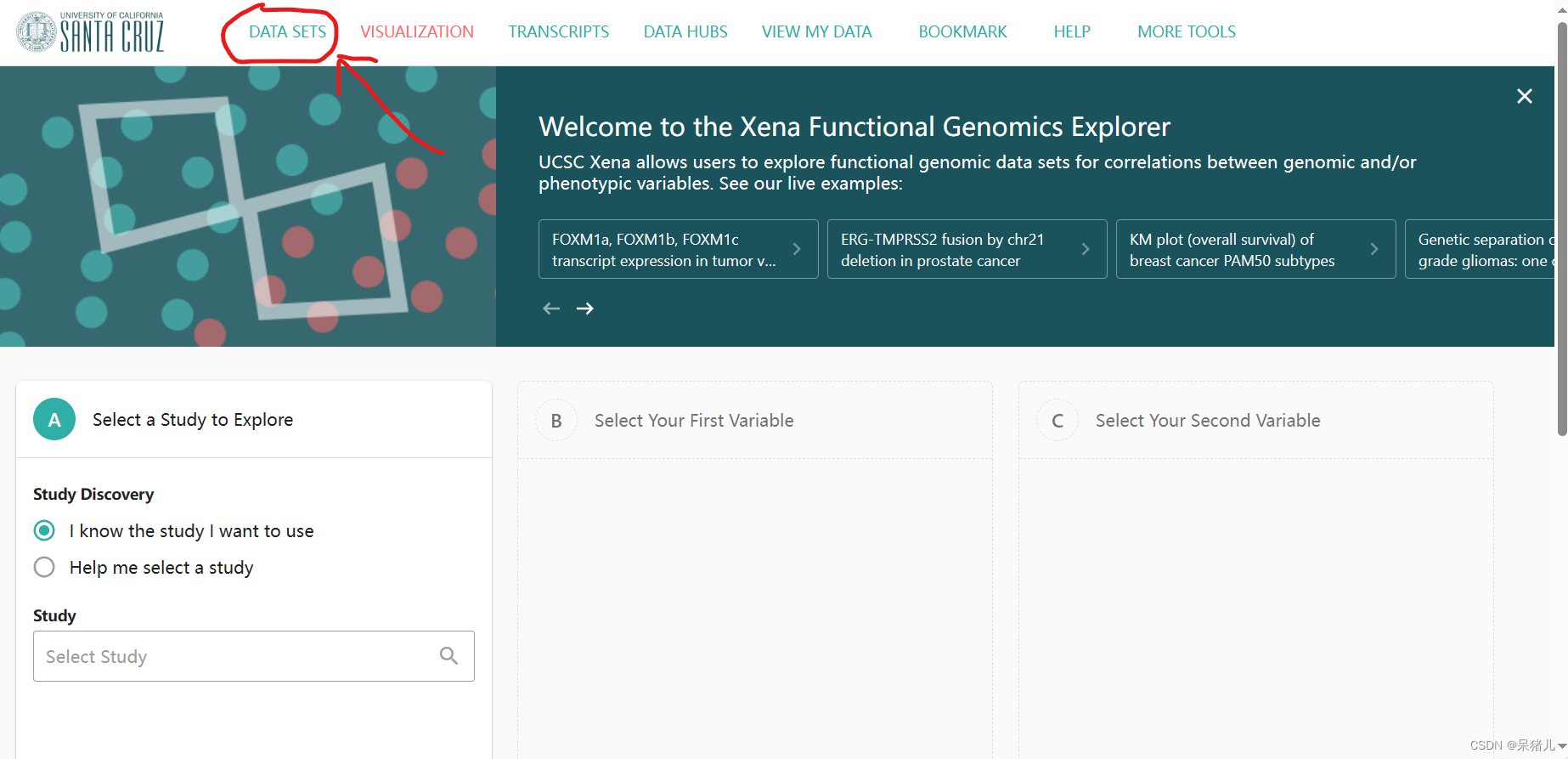
如下图所示为Xena中存储数据集的页面,其中(1)就是收录的各种疾病的数据集,(2)是筛选条件,由于该网站不仅收录了TCGA数据库的,同样还收录了其他数据库的数据集,因此可以根据自己的需要选择想要的数据库及数据集。我们想要下载是TCGA数据集,因此勾选GDC Hub 和TCGA Hub(至于为什么会选择这两个,在下面会有个人的一点理解和解释)
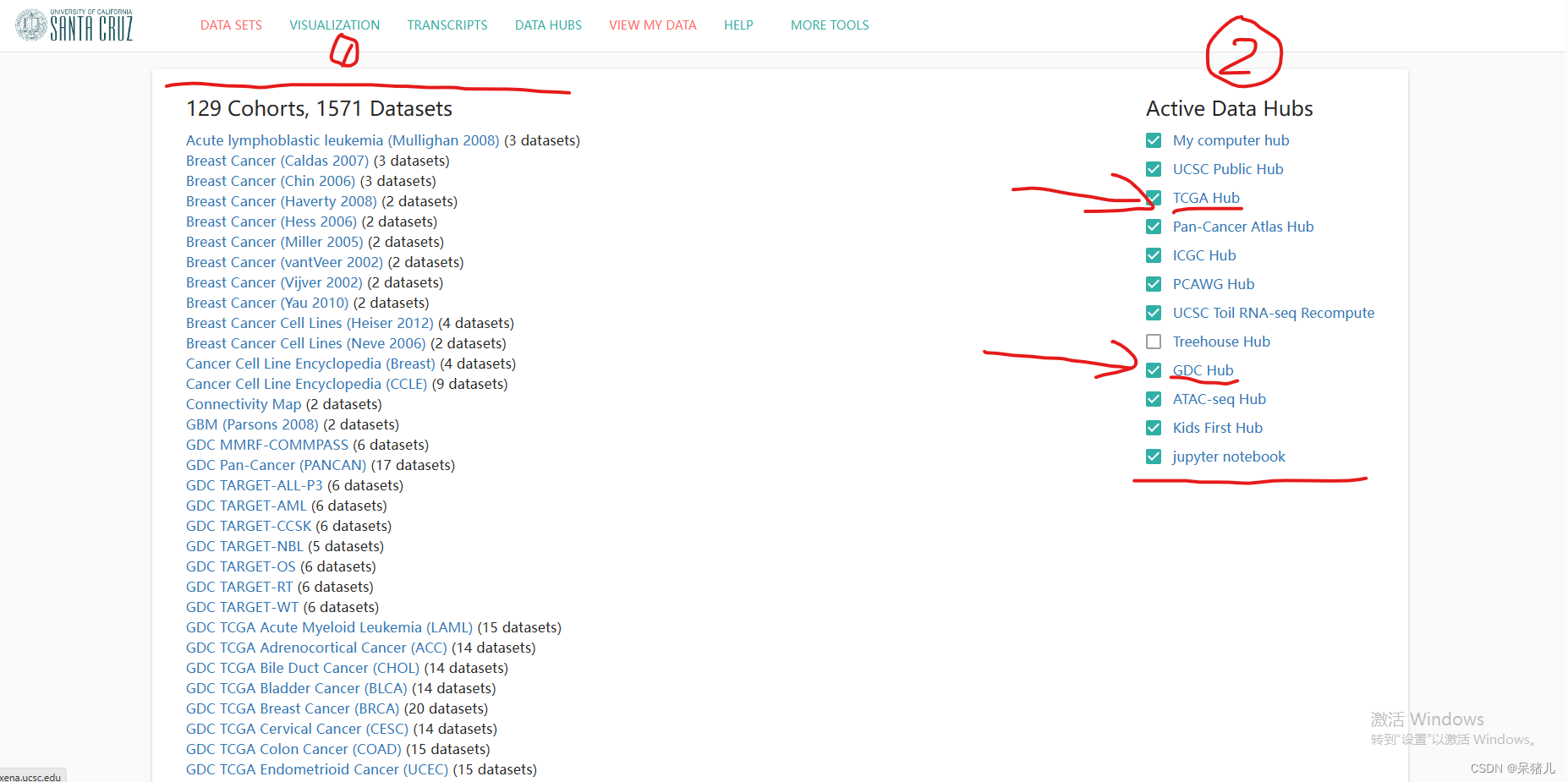
如下图所示,为勾选GDC Hub 和TCGA Hub的筛选情况,可以从图中看到两个都是TCGA数据集,那么区别是什么呢?
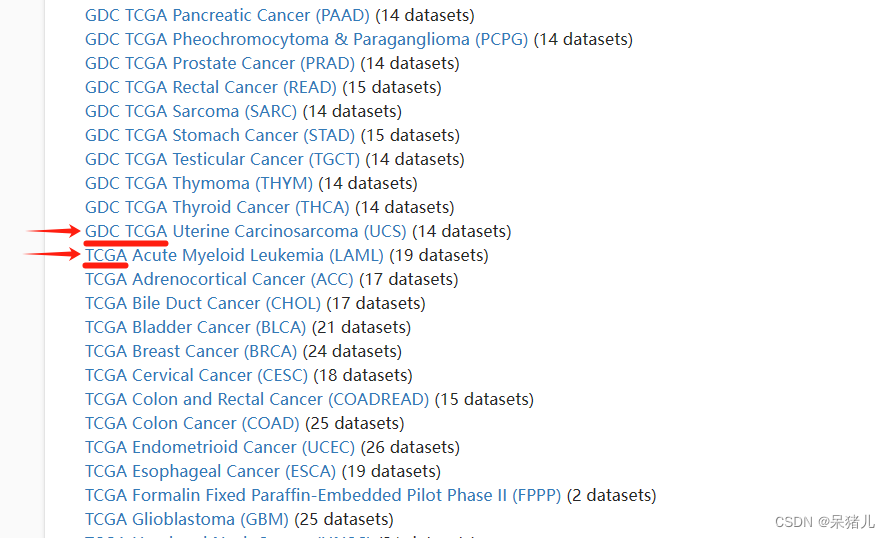
直接以本教程要展示的疾病——肺腺癌(lung adenocarcinoma,LUAD)为例,分别找到对应的GDC-TCGA数据集和TCGA数据集,如下所示,分别点进去可以看到详细信息。


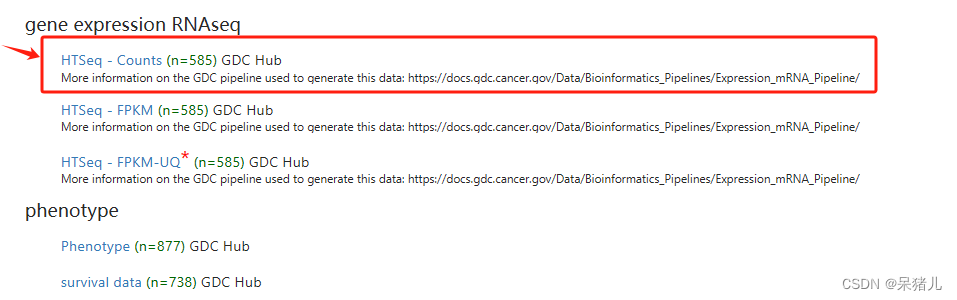
如下图所示为GDC-TCGA肺腺癌数据集,可以看到整个界面干净简洁,并且图中标注出了4个红字部分就是我们后续要用到的数据,分别是原始Count,FPKM,Phenotype,以及survival data。
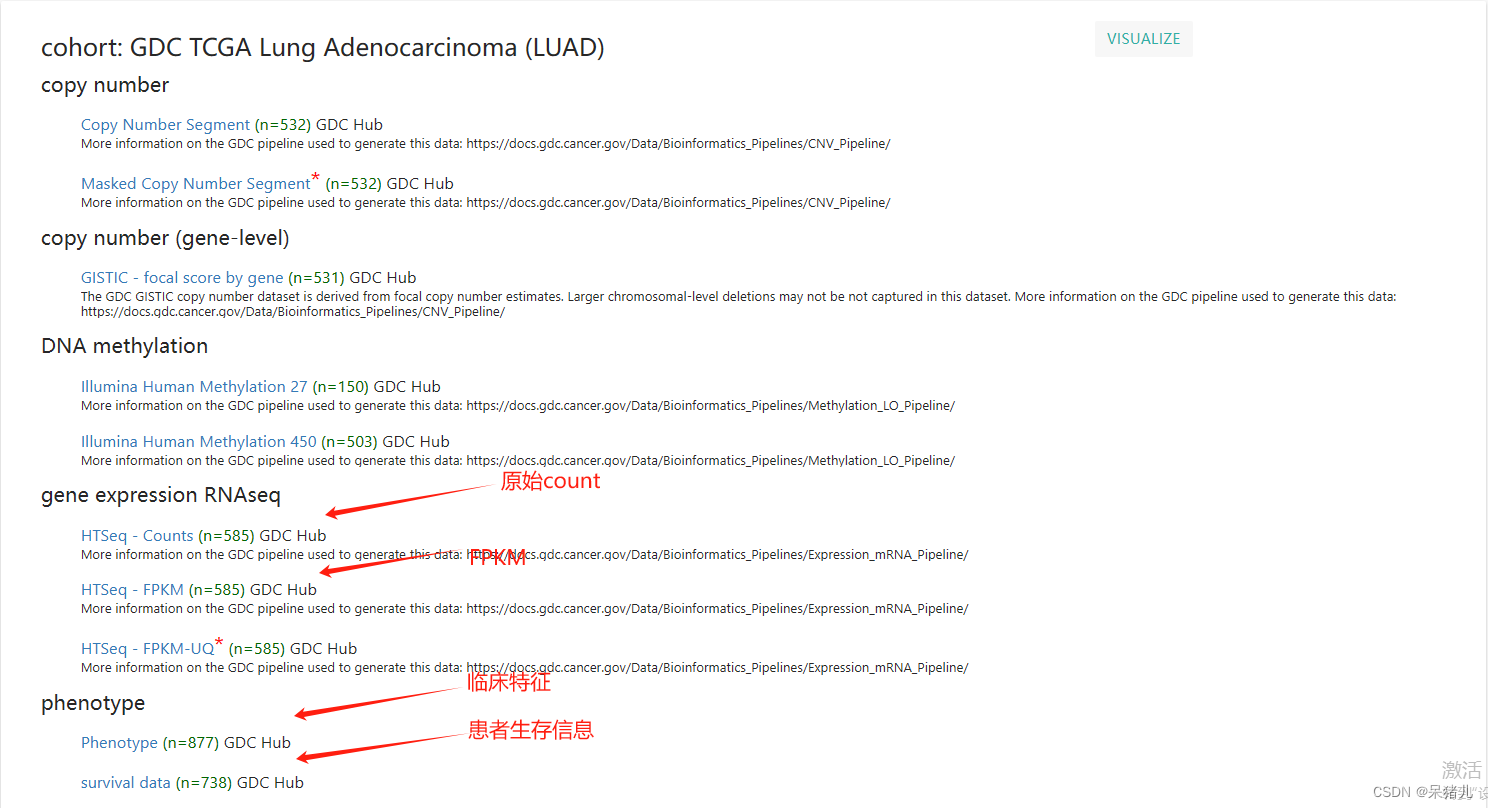
4个数据解释如下:
- counts——转录组的原始表达矩阵,里面对应的基因表达量又被称作raw_count,行名为基因symbol,列名为样本名(也是病人的id,可以将每一列看作一个病人)
- fpkm——raw_count经过转换后的表达矩阵,其计算公式可参考一文了解Count、FPKM、RPKM、TPM | 相互间的转化 | 收藏教程
- phenotype——病人的临床信息,包含分组信息,肿瘤分期,分级,年龄,性别等
- survival——病人的生存信息,里面通常会有4列信息,两列的病人的id,另外两列:OS——生存状态(0表示存活,1表示死亡),OS.time——生存时间
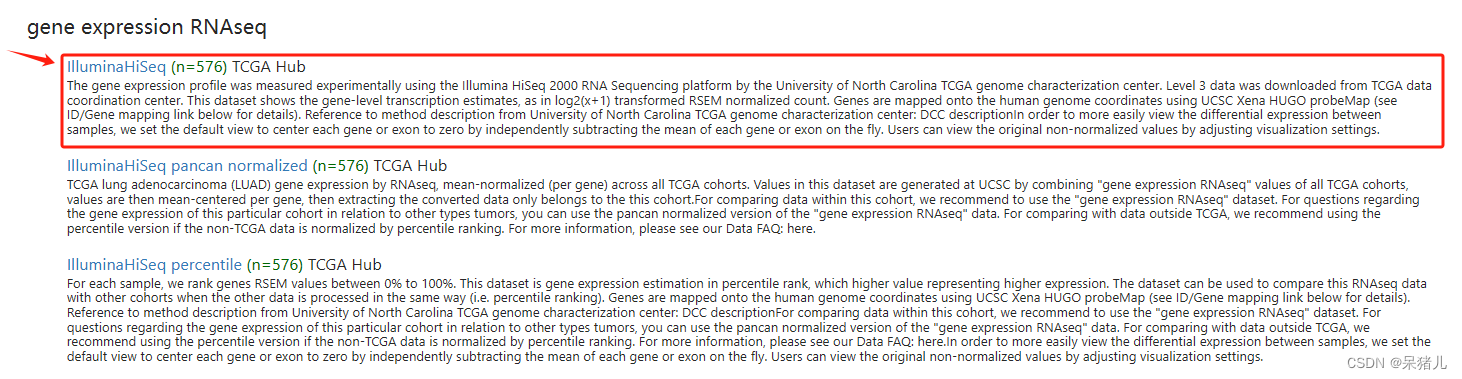
如下图所示为TCGA肺腺癌数据集,可以看到整个界面信息比较多,但也包括了基因表达量及患者的临床信息和生存信息。
那么这两者区别是什么?可参考别人的教程:UCSC Xena数据库中GDC TCGA数据和TCGA数据的区别和聊UCSC xena的数据下载问题,但是解释的都不是很清楚,并且官方解释也是晦涩难懂,对于后续分析该选择哪个并没有做出很好的解答。
总的来说就是TCGA这个数据集的发行年份更早,而GDC则是发行年份更晚一些,做科研的小伙伴也都知道一般都要追前沿,那我们这也不例外,选择一个最新的GDC数据库。
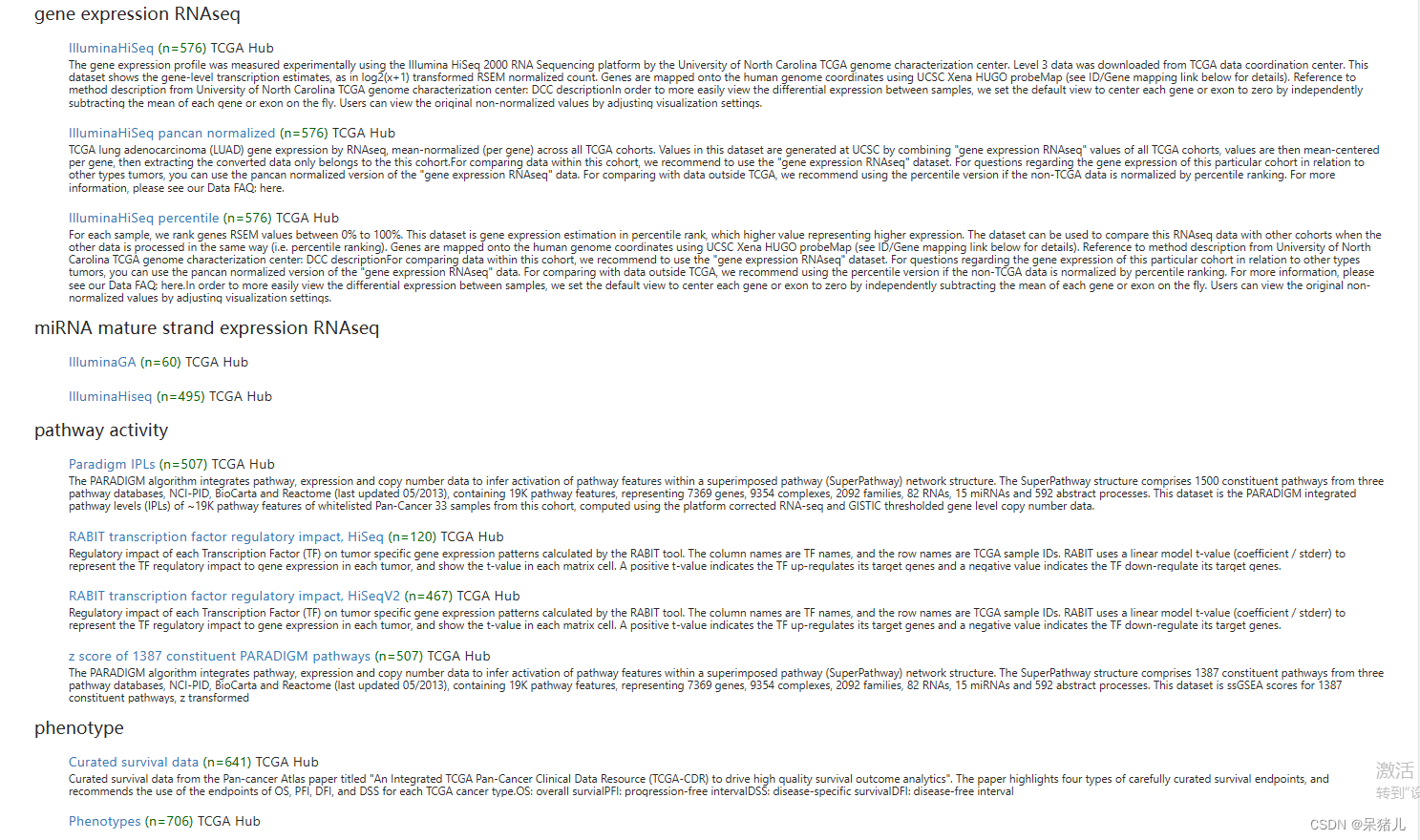
同时于偶然间发现了一件事:GDC的数据处理要比TCGA更加方便!如下图所示,分别点进去GDC中的Counts和TCGA中的IlluminaHiSeq。
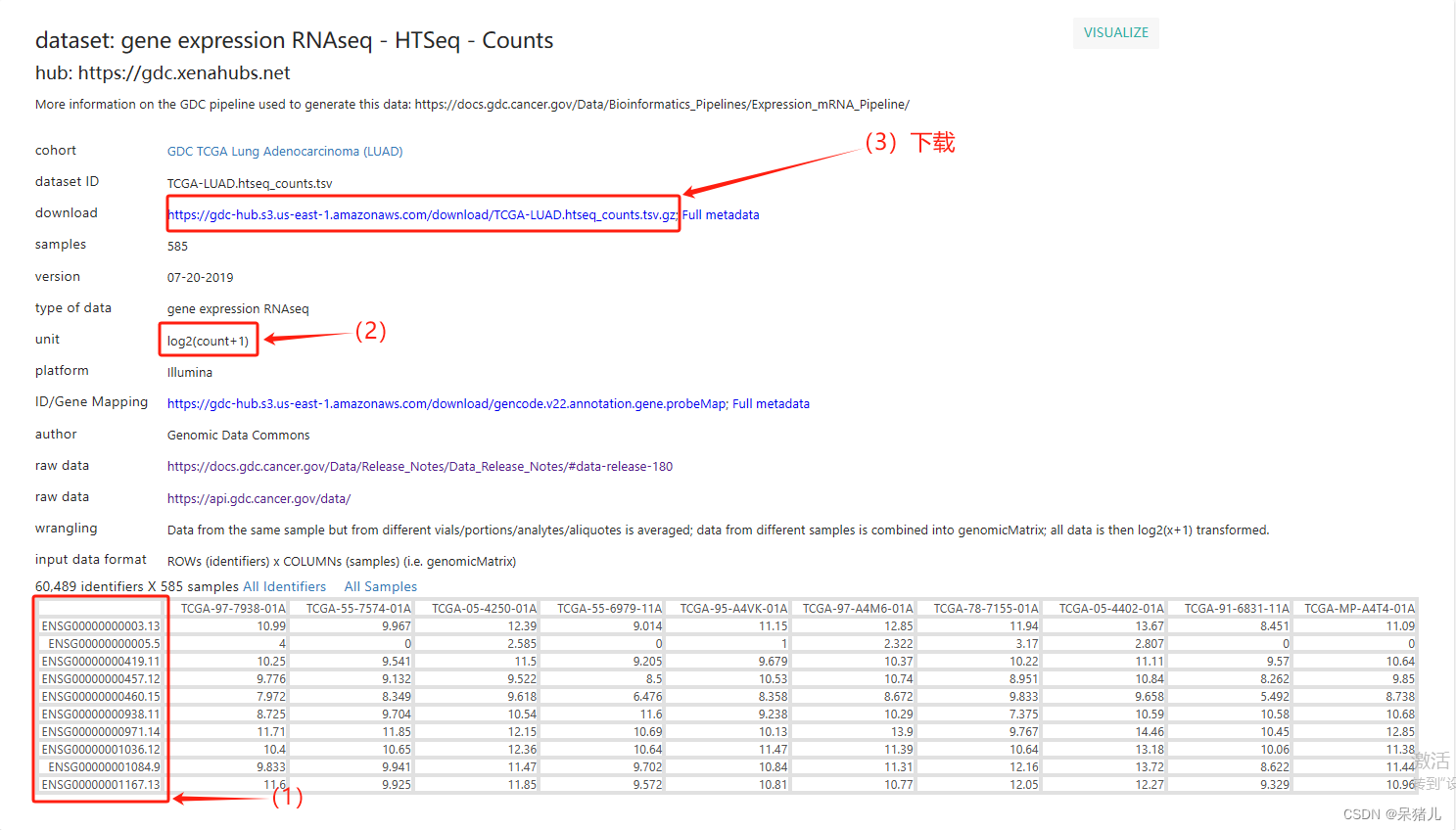
结果如下所示是GDC的数据,其中(1)是基因的ensemble_ID,这个玩意可以直接转换成基因symbol,(2)表示该数据集GDC数据库是进行了log2(X+1)处理的,因此后续处理是要反转一下才能获得想要的count数据,(3)是下载的链接,只要点击即可下载数据。
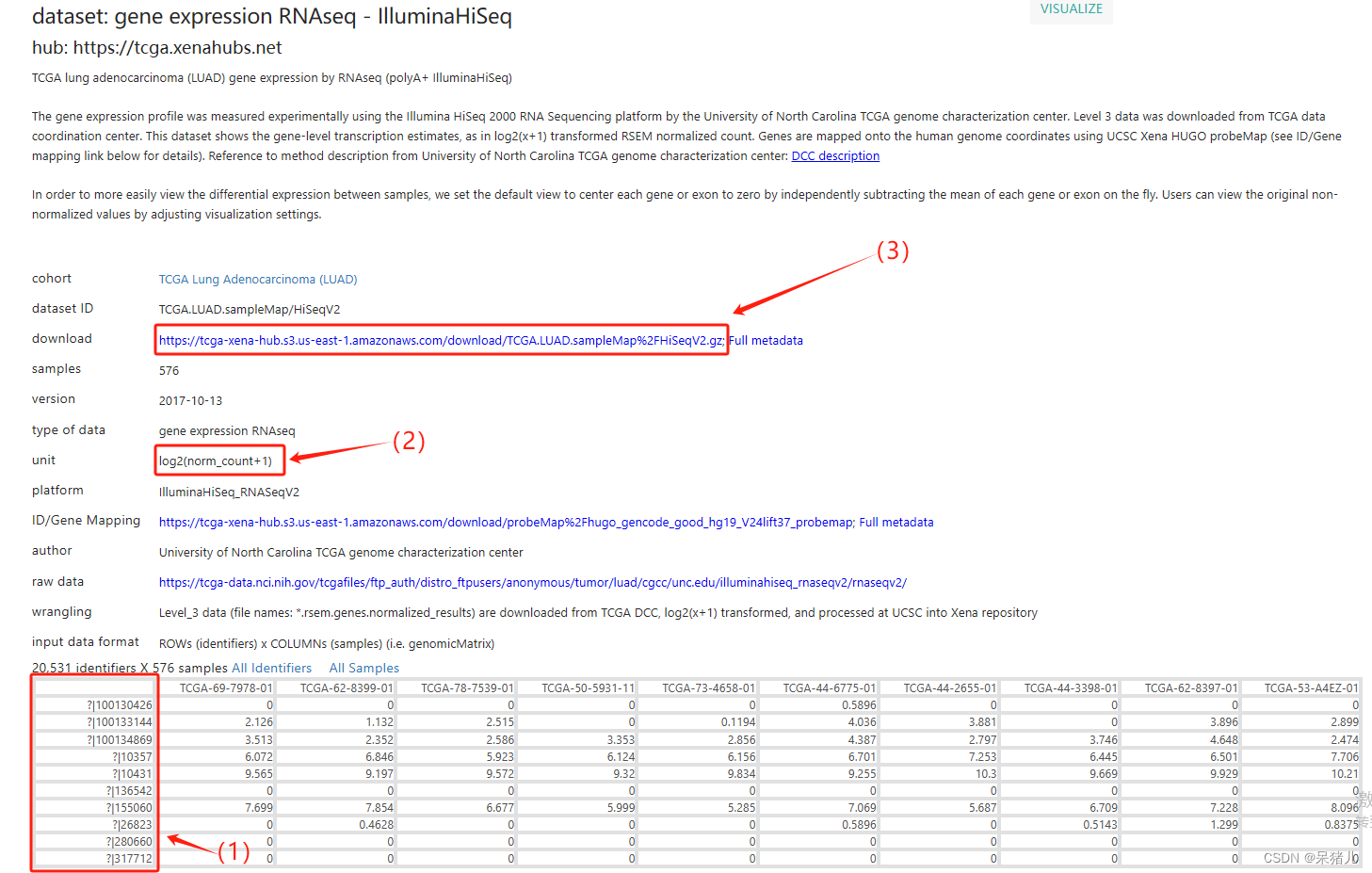
 TCGA的数据如下图所示,其中(1)对应的不知道是什么,推测应该是探针的ID(没深入研究),也有可能是发行年份较早有些基因缺失。但是这个玩意要想转换成基因symbol,应该需要先找到对应的文件,才能转换成对应基因symbol,同时细心的小伙伴也可以发现TCGA这个数据库他只有20531个基因,而前面GDC数据库则是60489个基因,(2)同样表示该数据集是进行了log2(X+1)处理的,因此后续处理也是要反转一下,(3)是下载的链接。
TCGA的数据如下图所示,其中(1)对应的不知道是什么,推测应该是探针的ID(没深入研究),也有可能是发行年份较早有些基因缺失。但是这个玩意要想转换成基因symbol,应该需要先找到对应的文件,才能转换成对应基因symbol,同时细心的小伙伴也可以发现TCGA这个数据库他只有20531个基因,而前面GDC数据库则是60489个基因,(2)同样表示该数据集是进行了log2(X+1)处理的,因此后续处理也是要反转一下,(3)是下载的链接。
总的来说已目前的理解来看GDC数据库中的数据集更加方便简洁一些,可以直接通过ensemble_ID对应基因,因此选用一个更加方便的方法——GDC数据库,前面我们也说了图中(3)是下载链接;“照猫画虎,依葫芦画瓢”,以同样的方式下载FPKM,Phenotype,以及survival data,下载完后注意后缀是不是XX.tsv.gz或XX.tsv。如下图所示是下载好的数据集们,接下来要在R中让他们展示自我并做进一步处理。
2. 数据处理(Rstudio)
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/XX/XX/XX') # 设置工作路径
if(!dir.exists('./00_rawdata')){
dir.create('./00_rawdata')
} # 判断该工作路径下是否存在名为00_rawdata的文件夹,如果不存在则创建,如果存在则pass
setwd('./00_rawdata/') # 设置路径到刚才新建的00_rawdata下
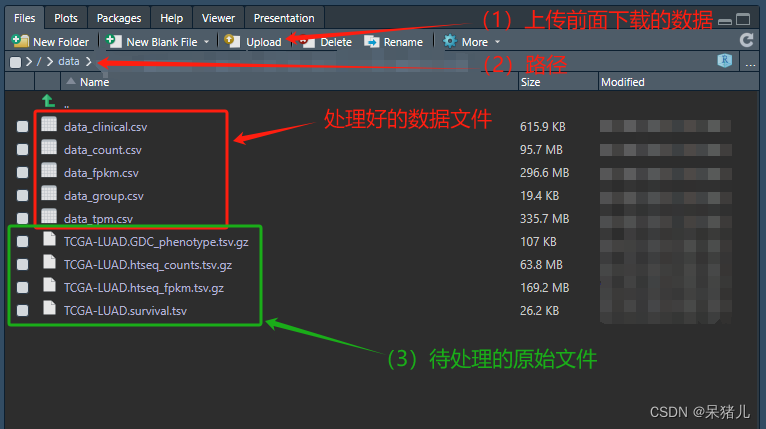
上传前面下载的4个tsv数据到Rstudio中(注意:最好上传到刚才创建的00_rawdata文件夹下),如下图所示,绿色的框就是要处理的原始文件。
首先加载要用的包,万能的tidyverse,dplyr,rtracklayer如果没有安装这3个包,可以通过install.packages(‘XXX’)指令安装,XXX就是包的名字,例如:install.packages('tidyverse')
library(tidyverse)
library(dplyr)
library(rtracklayer)
### 读入下载的数据
tcga_count <- read_tsv(file = './TCGA-LUAD.htseq_counts.tsv.gz') # count数据
tcga_fpkm <- read_tsv(file = "./TCGA-LUAD.htseq_fpkm.tsv.gz") # fpkm数据
tcga_survival <- read_tsv(file = "./TCGA-LUAD.survival.tsv") # 患者生存状态
tcga_phenotype <- read_tsv(file = "./TCGA-LUAD.GDC_phenotype.tsv.gz") # 患者临床信息
### count数据处理
tcga_count <- as.data.frame(tcga_count) # 将导入的文件转成数据框格式
tcga_count <- tcga_count[!duplicated(tcga_count$Ensembl_ID), ]
tcga_count <- column_to_rownames(tcga_count, var = 'Ensembl_ID') # 将文件第一列转为行名
tcga_count <- 2^tcga_count-1 # xena下载的数据经过了log2+1转化,需要将其还原
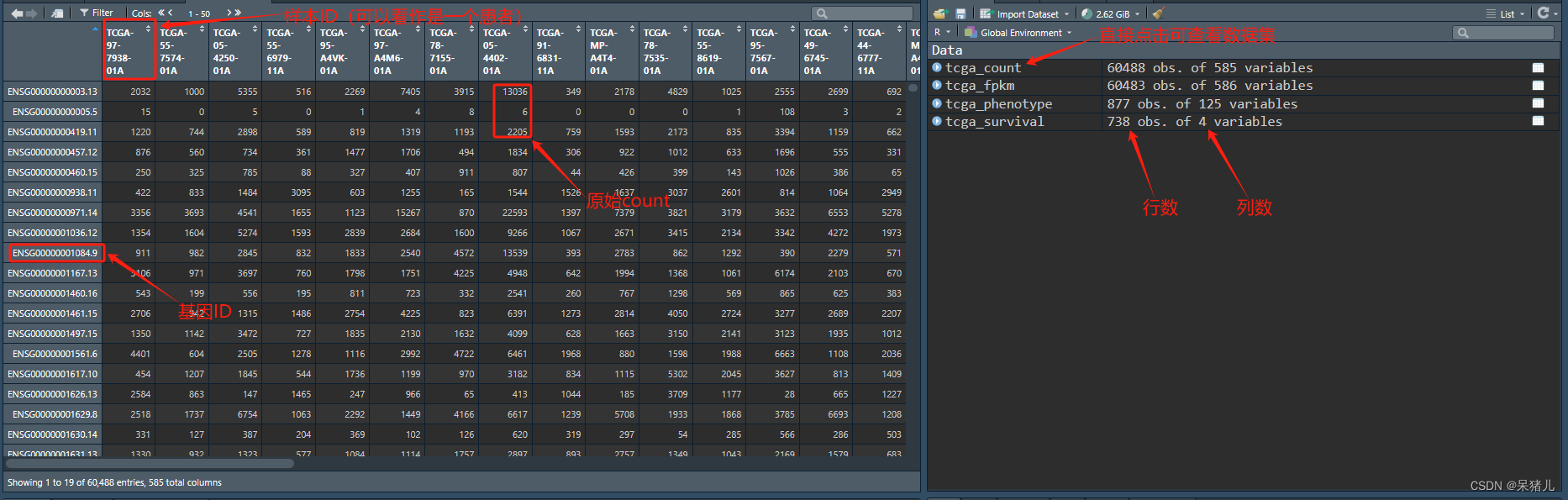
点击右上角的tcga_count可以查看数据集,这里要注意一下:要看一下count数据集中的原始count是不是都是整数,如果不是整数,可以看一下是否经过反转(2^tcga_count-1)。
接下来要导入一个比较重要的文件——gencode.v22.annotation.gtf
从bing上搜索gencode.v22.annotation,检索到的第一个就是基因注释文件
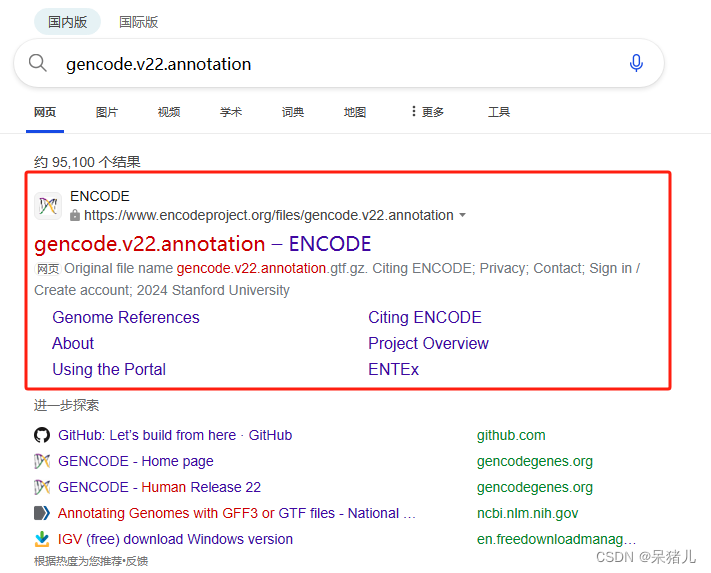
进入ENCODE的gencode.v22.annotation页面,直接点击download可下载。
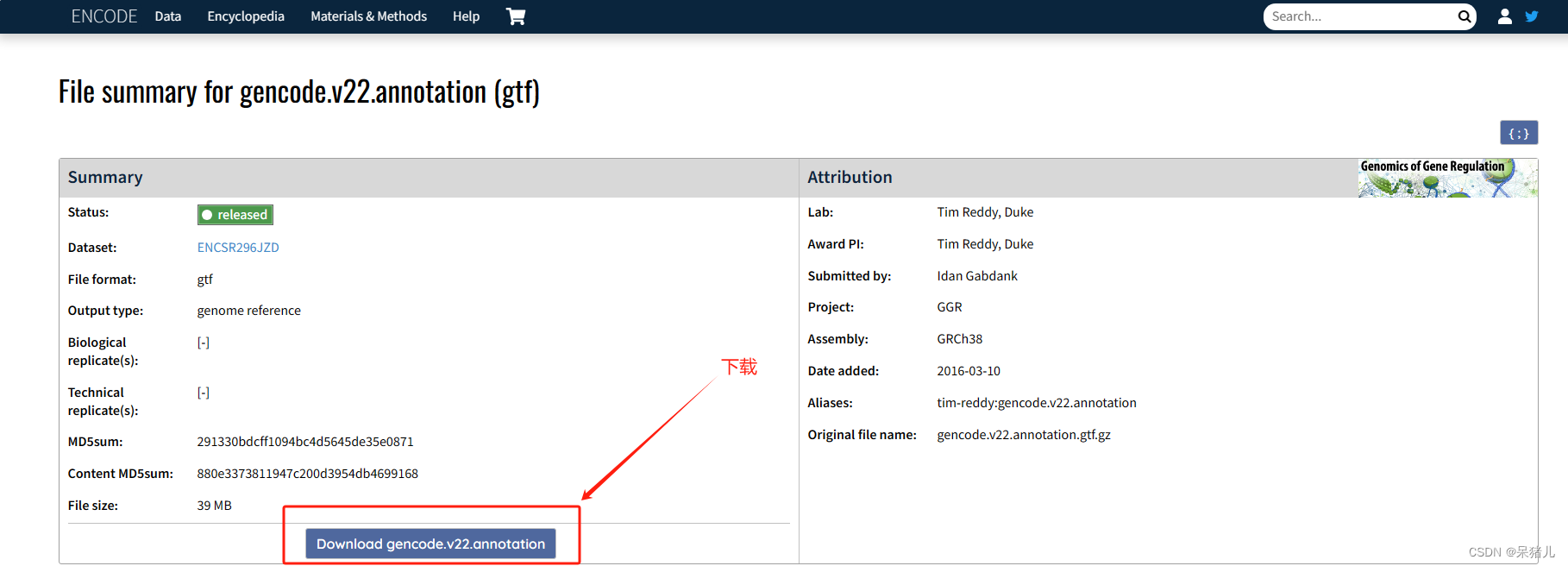
下载后是个.gz的压缩包,解压后同样上传到Rstudio的00_rawdata中,运行如下代码导入:
gene_annotation <- import('./gencode.v22.annotation.gtf.gz')
gene_annotation <- as.data.frame(gene_annotation)#将文件转换为数据框格式
gene_annotation <- gene_annotation [gene_annotation$type == 'gene', ] # 筛选为gene的类型
gene_annotation <- dplyr::select(gene_annotation, gene_id, gene_name, seqnames, start, end, width, strand, gene_type)
看看最后处理好的gene_annotation长啥样,点击Rstudio右上角的gene_annotation 如下图所示:
- gene_id——基因的ensemble_ID
- gene_name——基因名
- seqnames——基因位于哪条染色体
- start——该基因于染色体上的起始位置
- end——该基因于染色体上的终止位置
- width——该基因的长度(注意:凭借这个就可以计算FPKM)
- strand——链的方向,+表示正链,-表示负链(关于链的方向的描述信息可以自行查找资料,在这里跟教程无关不做过多介绍)
- gene_type——基因的类型,有的是miRNA,有的是lincRNA…
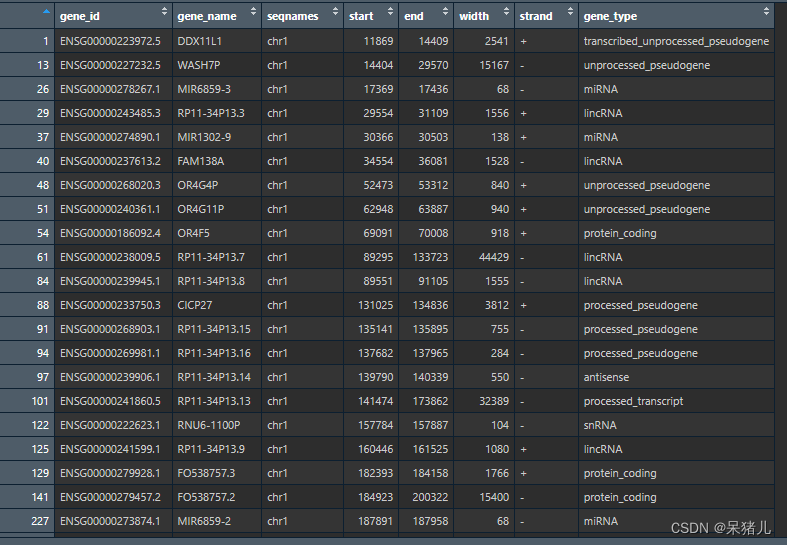
这里我们用到的是前两列gene_id和gene_name
gene_symbol <- gene_annotation[,(1:2)] # 筛选gene_annotation文件中的第一列和第二列
colnames(gene_symbol) <- c("ID", "symbol") # 将gene_symbol文件的列名改成ID和symbol
gene_symbol如下图所示,第一列是ensemble_ID,第二列是基因symbol
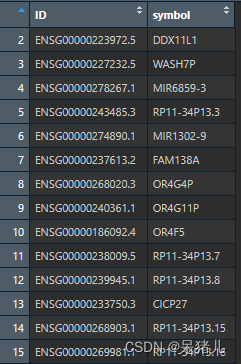
准备好前面的count数据和基因注释文件后,运行下面代码,每句代码后都有注释信息,可以挨个查看,比对处理前后的变化
data_tcga <- tcga_count
data_tcga$ID <- rownames(data_tcga) # 给data_tcga添加一列,列名为ID
data_tcga$ID <- as.character(data_tcga$ID) # 将data_tcga文件中ID列从数值类型数据转化为字符串类型数据
gene_symbol$ID <- as.character(gene_symbol$ID) # 将gene_symbol文件中ID列从数值类型数据转化为字符串类型数据
data_tcga <- inner_join(data_tcga, gene_symbol, by = "ID") # 将data_tcga文件和gene_symbol文件根据ID列进行合并(这样就能获得ID对应的基因名,但是都排在最后)
data_tcga <- dplyr::select(data_tcga, -ID) # 删除ID列
data_tcga <- dplyr::select(data_tcga, symbol, everything()) # 重新排列,将最后一列的基因名放到第一列(如果不加everything只会选择symbol一列)
data_tcga <- mutate(data_tcga, rowMean=rowMeans(data_tcga[grep('TCGA',names(data_tcga))])) # 添加一列为表达量的平均值
data_tcga <- arrange(data_tcga, desc(rowMean)) # 把表达量的平均值从大到小排序
data_tcga <- distinct(data_tcga, symbol, .keep_all = T) # distinct函数被用于去重,.keep_all参数表示去重后返回数据框的所有列向量
data_tcga <- dplyr::select(data_tcga, -rowMean) # 去除rowMean这一列
data_tcga <- tibble::column_to_rownames(data_tcga, var = "symbol") ## 把第一列变成行名并删除
dim(data_tcga)
上面的代码说白了就是一个去重加基因注释,结果如下图所示,行为基因symbol,列为样本名,但是同样每个样本后有个01A和11A这个是与癌症相关的,01-09为肿瘤,10-19为正常对照。
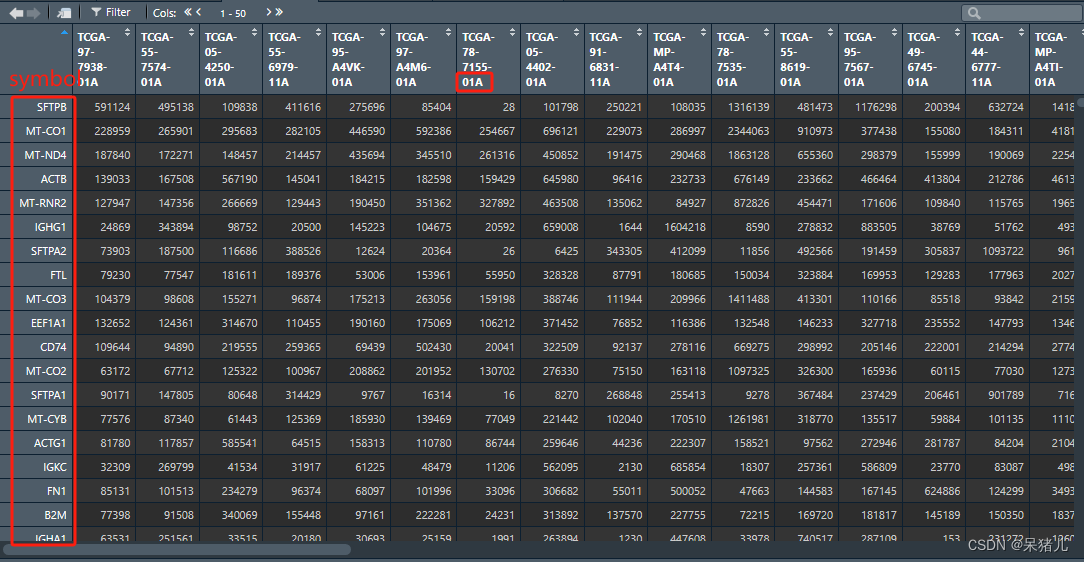
接下来根据样本最后的这个数字初步筛选癌症和对照样本
### 筛选癌症和对照样本。01-09为肿瘤,10-19为正常对照
mete <- data.frame(colnames(data_tcga))
for (i in 1:length(mete[, 1])) {
num <- as.numeric(as.character(substring(mete[i, 1], 14, 15))) # 提取每行第一列中第14,15字符
if(num %in% seq(1, 9)){mete[i, 2] = "T"} # 判断:如果提取的数字在0-9之内就在每行第二列加上T表示肿瘤样本
if(num %in% seq(10, 29)){mete[i, 2] = "N"} # 判断:如果提取的数字在10-29之内就在每行第二列加上N表示正常对照
}
colnames(mete) <- c("id", "group")
table(mete$group)
mete$group <- as.factor(mete$group) # 将mete中group列转为因子模式
mete <- subset(mete, mete$group == "T") # subset函数,从数据框中选择出group组为T的行
exp_tumor <- data_tcga[, which(colnames(data_tcga) %in% mete$id)] # 从大样本中选出组为T的样本
exp_tumor <- as.data.frame(exp_tumor)
exp_tumor <- exp_tumor[, colnames(exp_tumor) %in% tcga_survival$sample] # 进一步筛选具有生存信息的样本
exp_control <- data_tcga[, which(!colnames(data_tcga) %in% mete$id)] # 反向从大样本中选出组为N的样本
exp_control <- as.data.frame(exp_control)
exp_control <- exp_control[, colnames(exp_control) %in% tcga_survival$sample] # 进一步筛选具有生存信息的样本
data_final <- cbind(exp_control, exp_tumor) # 将肿瘤样本文件和正常样本文件合并
data_final <- na.omit(data_final) # 去除含有NA的行
虽然已经根据样本ID可以区分肿瘤样本和正常样本,但是为了确保分组可靠,接下来依据前面导入的临床信息(tcga_phenotype)进一步确认分组
table(tcga_phenotype$sample_type.samples) # 查看样本类型,一共四种:FFPE Scrolls,Primary Tumor,Recurrent Tumor,Solid Tissue Normal,这里面我们选择Primary Tumor——原发肿瘤和Solid Tissue Normal——正常组织
table(tcga_phenotype$primary_site) # 查看癌症发生的部位
clinical_data <- subset(tcga_phenotype, sample_type.samples == 'Primary Tumor' | sample_type.samples == 'Solid Tissue Normal')
clinical_data <- clinical_data[clinical_data$submitter_id.samples %in% colnames(data_final), ]
clinical_data <- clinical_data[order(clinical_data$sample_type.samples, decreasing = T), ]
Group <- data.frame(sample = clinical_data$submitter_id.samples,
group = clinical_data$sample_type.samples)
Group$group <- ifelse(Group$group == 'Solid Tissue Normal', 'control', 'disease')
data_final <- subset(data_final, select = Group$sample) # 确保前面整理的count数据与整理的分组信息的样本是一致的
table(Group$group) ##control:59 disease:511
如下图所示是整理好的分组信息,第一列是样本的ID,需要和前面整理好的count数据集中的样本ID对应,第二列为样本所属的分组
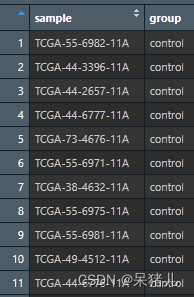
目前我们已经得到了整理好的count数据以及样本的分组信息,目前还差样本对应的生存信息和fpkm表达矩阵,样本的生存信息比较好处理,如下图所示为导入的样本生存信息,第一列和第三列都是样本ID,第二列OS是患者的生存状态,0表示存活,1表示死亡,第四列OS.time是患者的生存时间。
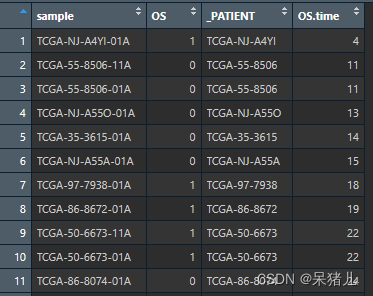
此时只需要去除掉第三行并且让生存信息表中患者的ID和前面整理好的分组信息表中的患者ID做个匹配即可,最后将整理好的count,group,以及生存信息表保存成csv格式即可。
tcga_survival <- tcga_survival[, -3]
tcga_survival <- tcga_survival[tcga_survival$sample %in% Group$sample, ]
write.csv(tcga_survival, './data_survival.csv')
write.csv(clinical_data, './data_clinical.csv')
write.csv(data_final, file = './data_count.csv')
write.csv(Group, file = './data_group.csv')
最后一步就是处理fpkm数据,这是因为在后续分析中除了差异表达分析会用到count数据,其余很多情况用的都是fpkm数据。处理的代码如下,其实整体思路和前面处理count时差不多,最后将结果保存为.csv形式即可。
### fpkm数据整理
tcga_fpkm <- as.data.frame(tcga_fpkm)
tcga_fpkm <- tcga_fpkm[!duplicated(tcga_fpkm$Ensembl_ID), ]
tcga_fpkm <- column_to_rownames(tcga_fpkm, var = 'Ensembl_ID')
tcga_fpkm <- 2^tcga_fpkm-1
dat_fpkm <- tcga_fpkm
dat_fpkm$ID <- rownames(dat_fpkm)
dat_fpkm$ID <- as.character(dat_fpkm$ID)
gene_symbol$ID <- as.character(gene_symbol$ID)
dat_fpkm<-dat_fpkm %>%
inner_join(gene_symbol,by='ID')%>%
select(-ID)%>% ## 去除多余信息
select(symbol,everything())%>% ## 重新排列
mutate(rowMean=rowMeans(.[grep('TCGA',names(.))]))%>% ## 求出平均数
arrange(desc(rowMean))%>% ## 把表达量的平均值从大到小排序
distinct(symbol,.keep_all = T)%>% ## symbol留下第一个
select(-rowMean)%>% ## 反向选择去除rowMean这一列
tibble::column_to_rownames(colnames(.)[1]) ## 把第一列变成行名并删除
dim(dat_fpkm)
dat_fpkm <- dat_fpkm[rownames(data_final), ]
dat_fpkm <- dat_fpkm[,colnames(data_final)]
dat_fpkm <- na.omit(dat_fpkm)
dat_fpkm <- log2(dat_fpkm + 1)
write.csv(dat_fpkm, file = './data_fpkm.csv')
到目前为止我们一共得到了5个文件,count,临床信息,分组信息,生存信息,以及最后的fpkm。
注:做到这一步,可以说是生信分析已经完成一半了,后续所有的分析都会基于前面处理的这几个文件,因此这最初的第一步也是最重要的一步,只有基础打好,后面分析才会可靠一些。
结语:
以上就是TCGA数据下载及处理的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。
如果觉得本教程对你有所帮助,点赞关注不迷路!!!
配套资源链接:配套资源(代码+原始数据+处理好的数据)
下期预告: 零基础入门转录组分析——数据处理(GEO数据库)
- 目录部分跳转链接:零基础入门生信数据分析——导读

















 1765
1765











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











