







硅的能带和态密度计算
根据VASP官方网站给的例子,计算立方金刚石型硅的能带和态密度
官方网站:https://www.vasp.at/tutorials/latest/bulk/part2/
b站视频:挑战第一个VASP计算:硅的能带和态密度【凝聚态计算入门】
学习笔记,在此记录
主要步骤:
1、获取到硅的结构信息(POSCAR)
2、进行结构弛豫,找出最佳晶格常数
3、使用含有最佳晶格常数的POSCAR进行自洽计算(静态计算),使用vaspkit生成态密度文件,使用originpro绘图
4、使用第一步或第二步生成的电荷密度文件和含有最佳晶格常数的POSCAR,修改KPOINTS为高对称点路径,再重新静态计算一遍,求得的本征值就是能带。使用vaspkit生成能带文件,使用originpro绘图
一、结构弛豫
1.1、官网解读
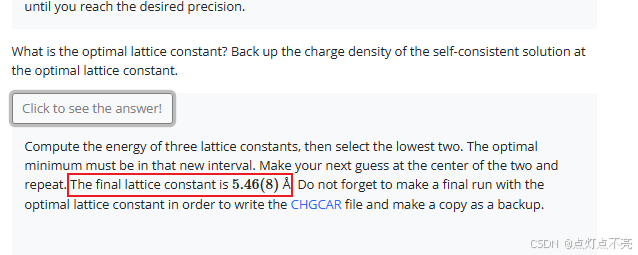
给出不同的晶格常数分别计算,找出使结构弛豫后能量最低的晶格常数
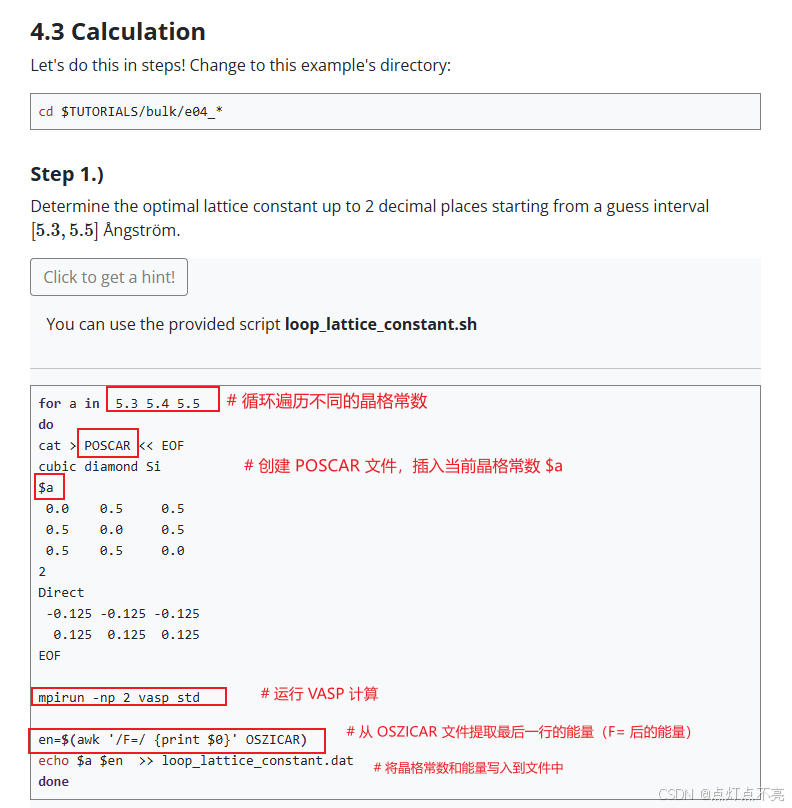
官网建议寻找最佳晶格常数的范围是【5.3,5.5】,步进为0.01,使用Python脚本进行了计算
1.1.1、输入文件
官网给出的输入文件 (POTCAR是硅的赝势文件)
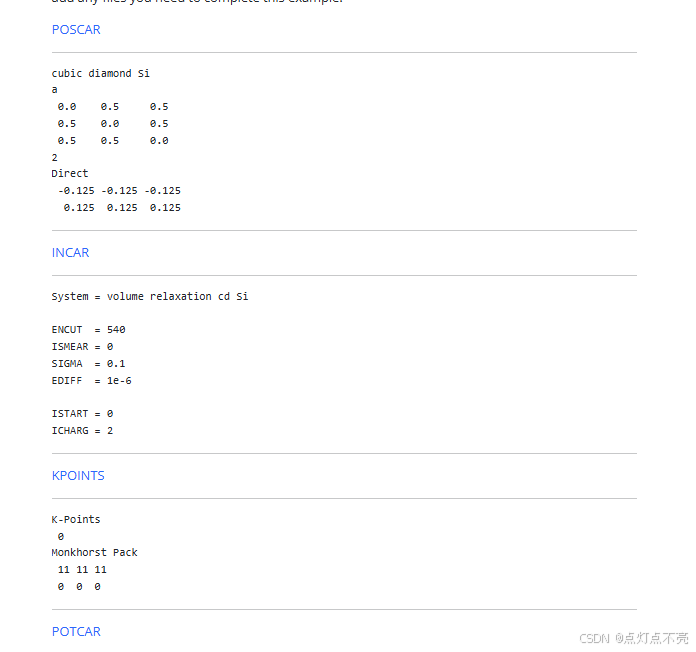
1.1.2、脚本计算
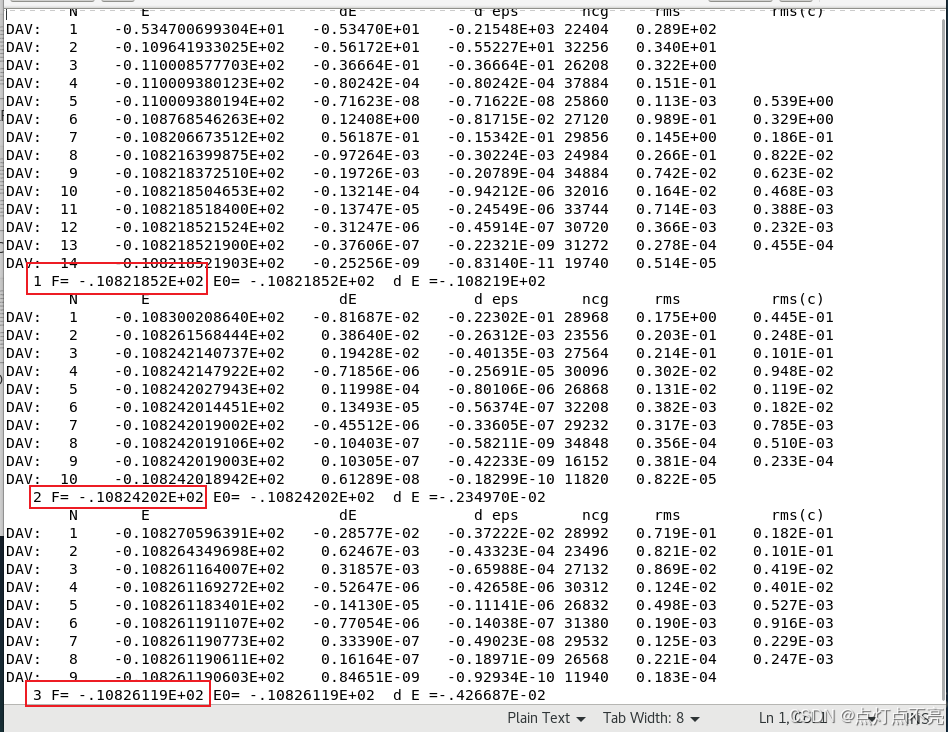
1.1.3、OSZICAR
OSZICAR 是 VASP(Vienna Ab-initio Simulation Package)中的一个输出文件,记录了自洽场计算过程中每一轮(迭代)的关键信息,主要包括总能量、能量变化、电子密度的收敛情况等。该文件的内容通常包括以下几部分:
每个迭代步骤的能量信息,通常包括:
-
F=:当前的总能量。 -
E0=:当前的总能量(通常与F=值相同)。 -
dE:当前步的能量变化。 -
d eps:能量变化的收敛度。 -
ncg:迭代的步数。 -
rms:残差误差。 -
rms(c):电子密度收敛的误差。这里展示一下OSZICAR
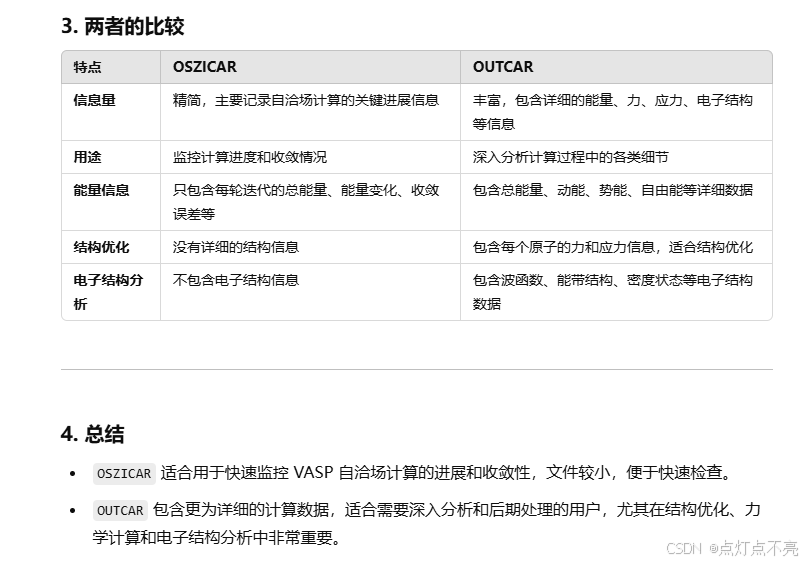
OSZICAR和OUTCAR对比
1.1.4、作图
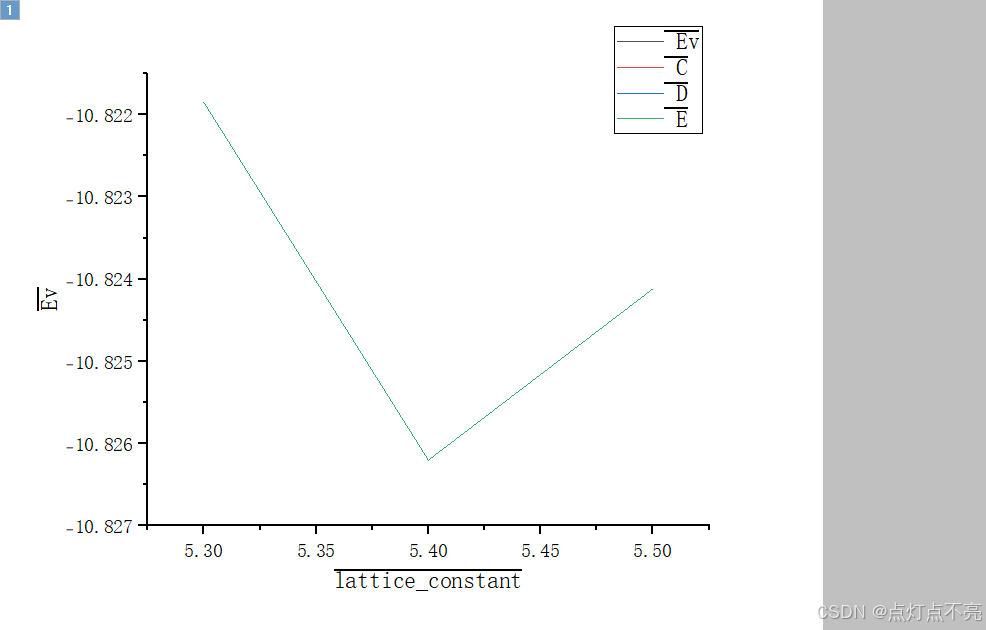
最后答案是晶格常数为 5.46(8) Å.时,结构收敛(能量最低)这里我没有进行计算,假设进行了计算,则最后的loop_lattice_constant.dat文件应该是这样:
lattice_constant Ev
5.3 -10.821852
5.4 -10.826202
5.5 -10.824119
用originpro作图
1.2、自行计算
1.2.1、POSCAR
使用官方例程的POSCAR,网上搜索硅的晶格常数为5.43Å,这里直接用。
cubic diamond Si #名称
5.43#晶胞参数(晶格常数,这里假设为5.43)
0.0 0.5 0.5#晶胞矢量a
0.5 0.0 0.5#晶胞矢量b
0.5 0.5 0.0#晶胞矢量c
2#原子数量
Direct
-0.125 -0.125 -0.125 #原子坐标
0.125 0.125 0.125
新建文件夹1_re,在1_re里新建文件POSCAR
1.2.2、INCAR
使用vaspkit生成INCAR
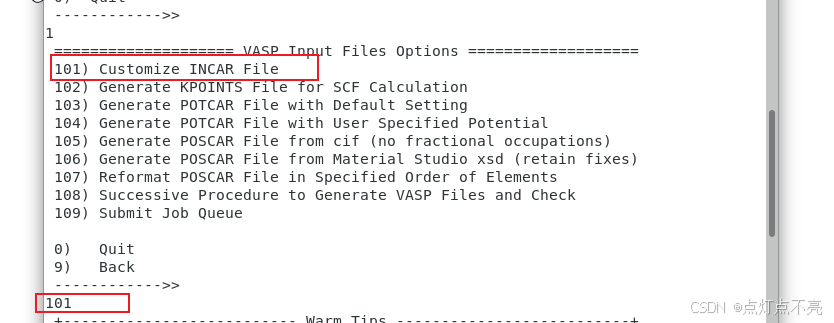
进入生成的INCAR

INCAR修改
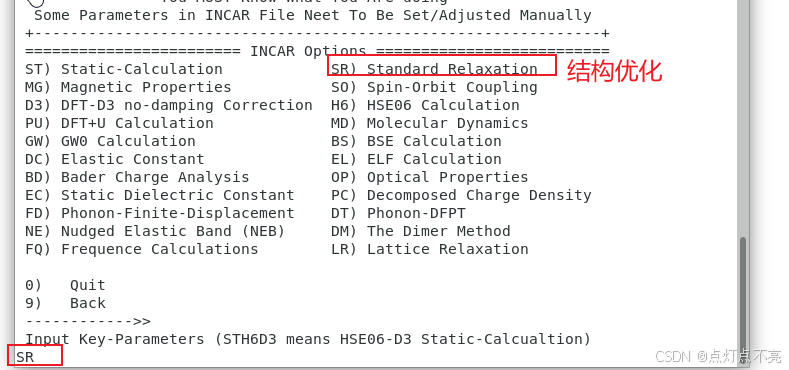
主要修改ISIF参数设置值
我们的INCAR和官网的INCAR区别在于:
1、官网INCAR ISIF参数为默认的0,即不设置 ISIF 并只改变晶格常数,进行结构弛豫,能量会发生变化。但这是因为 ISIF=0 不会自动优化晶格常数,也不会优化原子位置。在这种情况下,能量的变化主要是由于外部改变了晶格常数,导致结构应力不平衡,从而引起能量的改变。
2、我们的INCAR ISIF参数设置为3,是因为POSCAR中的晶格常数5.43Å是在网上找的,不一定准确,但5.43Å一定是在最佳晶格常数附近,并且我们本次侧重点是态密度和能带,不希望在寻找最佳晶格常数消耗太多时间,所以设置ISIF为3,即同时优化晶格常数和原子位置。这样,VASP 会在结构优化过程中同时优化晶格常数(体积)和原子位置,从而找到最低能量的结构。
3、ISIF参数设置为3是有风险的,对于二维体系(2D材料、切片等),不能用 ISIF=3,因为大量机时会被浪费在真空层优化
4、当双变量优化,可先ISIF=2(用实验参数) 优化原子坐标,然后ISIF=3优化晶胞;检查Pulay Stress
5、ISIF=3优化晶胞时出现了明显的能量振荡 或晶胞显著变形,就应该立即停下。
vasp手册查询ISIF参数
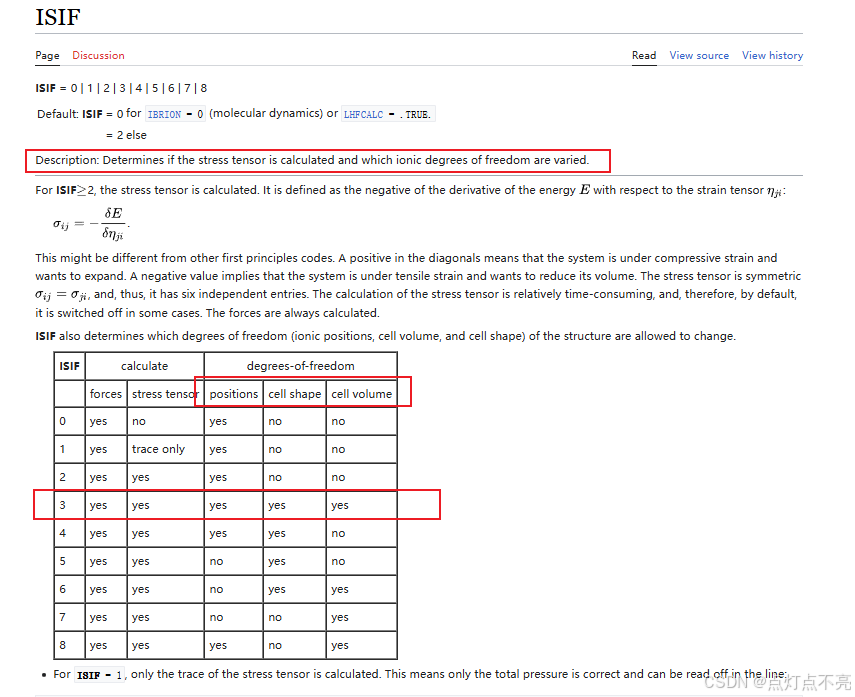
ISIF决定了原子的位置、形状、体积是否参与了计算
vasp手册查询网站:https://www.vasp.at/wiki/index.php/ISIF
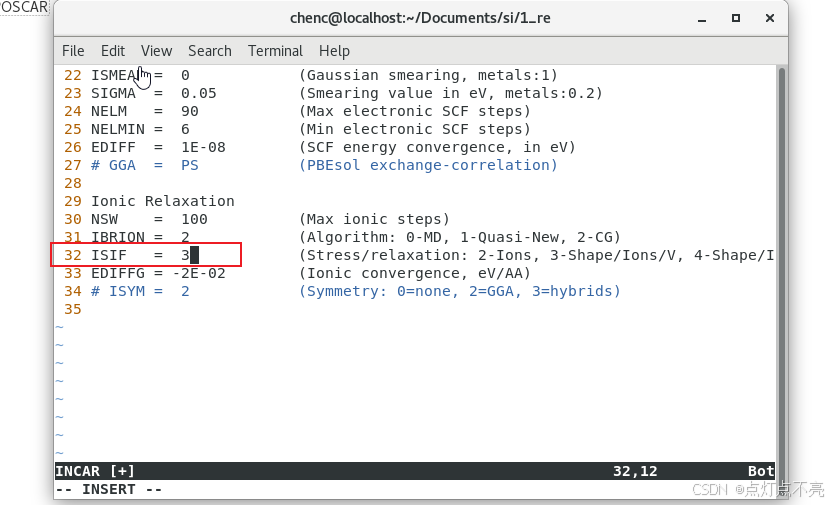
修改INCAR,ISIF设置为3
1.2.3、KPOINTS
打开vaspkit,获得KPOINTS
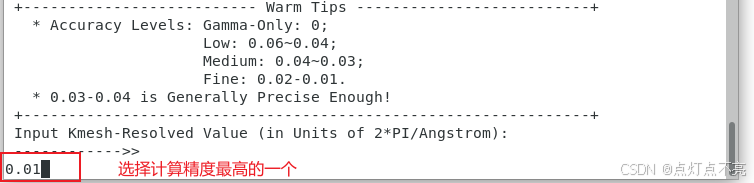
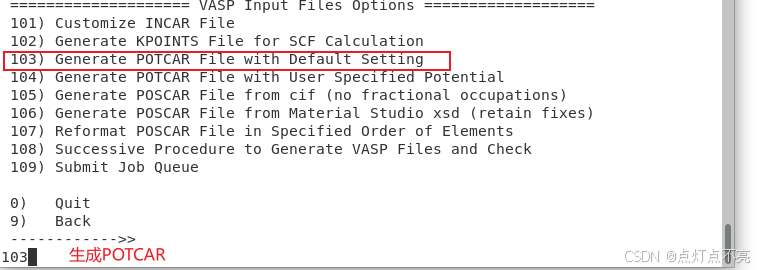
1.2.4、POTCAR
一般情况下,如果POSCAR格式正确,在上一步使用vaspkit生成KPOINTS的同时会自动生成POTCAR
如果没有自动生成POTCAR,再次打开vaspkit,获得POTCAR
报错信息:未能识别POSCAR中的元素

POSCAR文件格式注意:

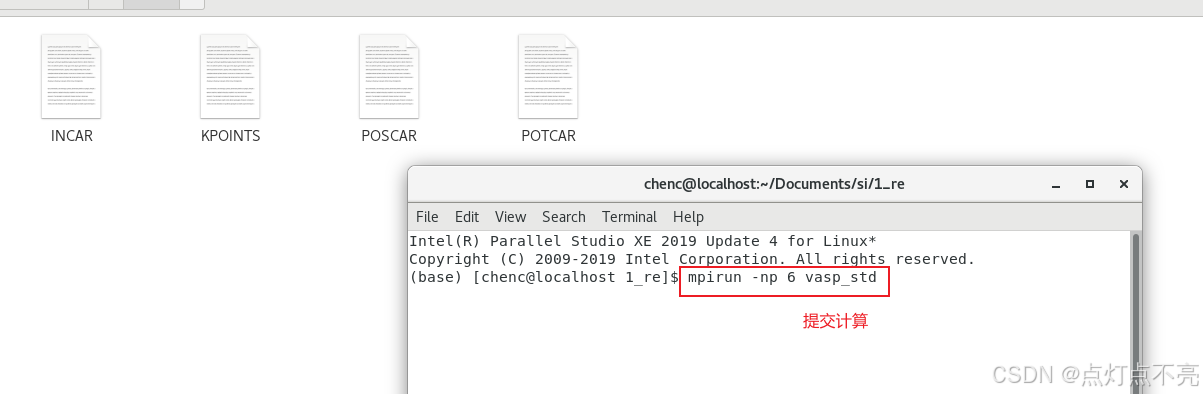
重新修改POSCAR后,获得POTCAR。至此,四个输入文件全部准备好了
1.2.5、提交计算
计算完成后打开CONTCAR(计算后的结构),对比计算前的结构(POSCAR)
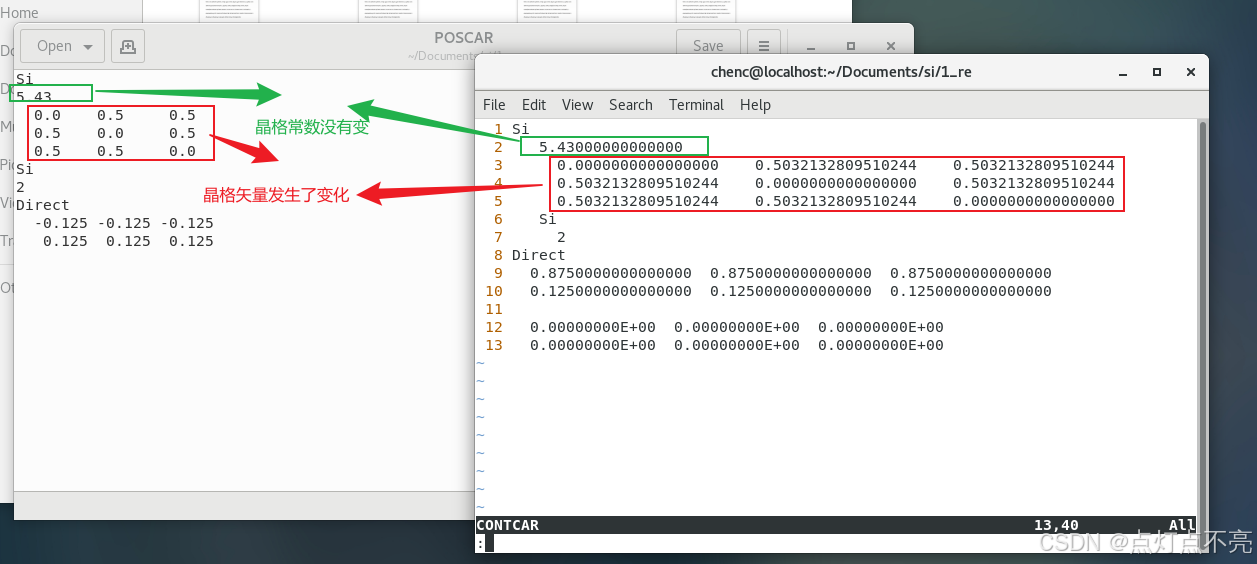
晶格常数定义:晶格常数(Lattice Constant)是描述晶体结构的基本参数之一,指的是晶体中单位格点之间的距离。它定义了晶体的几何形状和周期性,是晶体学中非常重要的物理量。
可以发现,晶格矢量发生了变化(体积发生了变化)但晶格常数没变,所以要求出新的晶格常数
新的晶格常数 = 旧的晶格常数*变化后的矢量/变化前的矢量
新的晶格常数 = 5.43*0.5032132809510244/0.5 = 5.464896231128125
1.2.6、结果
最终我们得到结构弛豫后晶格常数为 5.464896231128125,这与官网给出的最佳值参考相差不大
二、计算态密度(自洽计算)
2.1、官网解读
2.1.1、输入文件
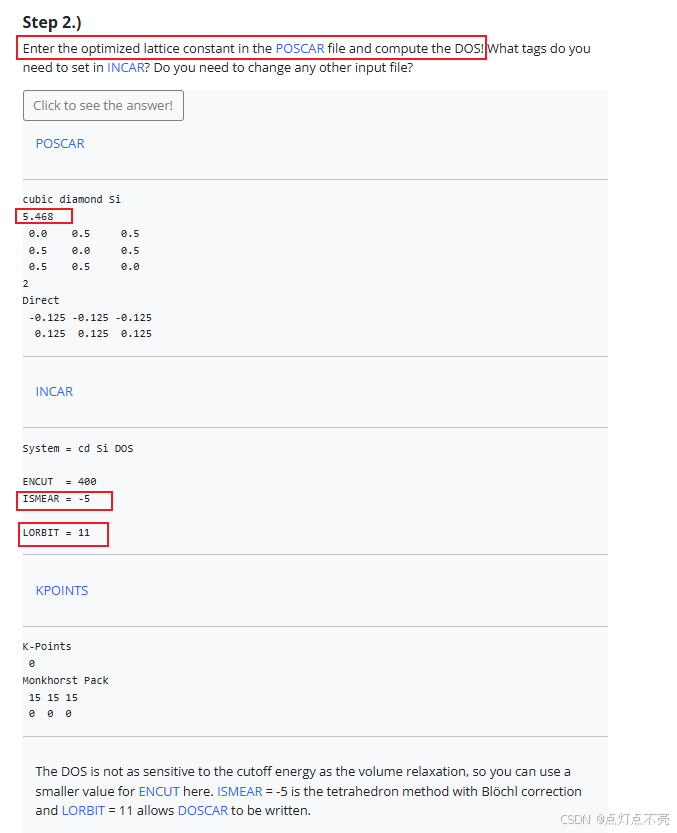
1、将上一步计算出的最佳晶格常数应用到POSCAR
2、DOS不像体积弛豫那样对截止能量敏感,因此您可以在这里对ENCUT使用较小的值。ISMEAR=-5是具有Blöchl校正的四面体方法(使DOS图毛刺更少)
3、LORBIT=11允许写入DOSCAR。
2.1.2、作图
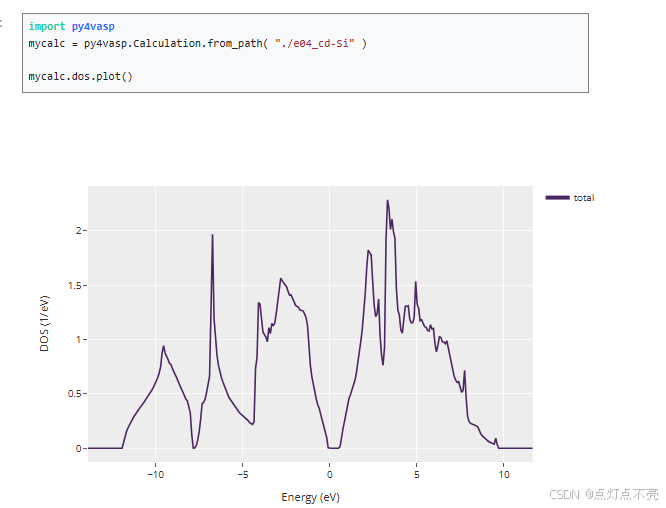
完成计算后得到TDOS.dat文件进行绘图
2.2、自行计算
2.2.1、POSCAR
将第一步经过结构弛豫后的结构文件(CONTCAR)拷贝一份作为POSCAR继续进行计算
这里有官网有区别的是:由上一步可知CONTCAR里晶格矢量发生了变化(体积发生了变化)但晶格常数没变,所以这里晶格常数是否修改都可以,因为格矢量发生了变化。
Linux命令:
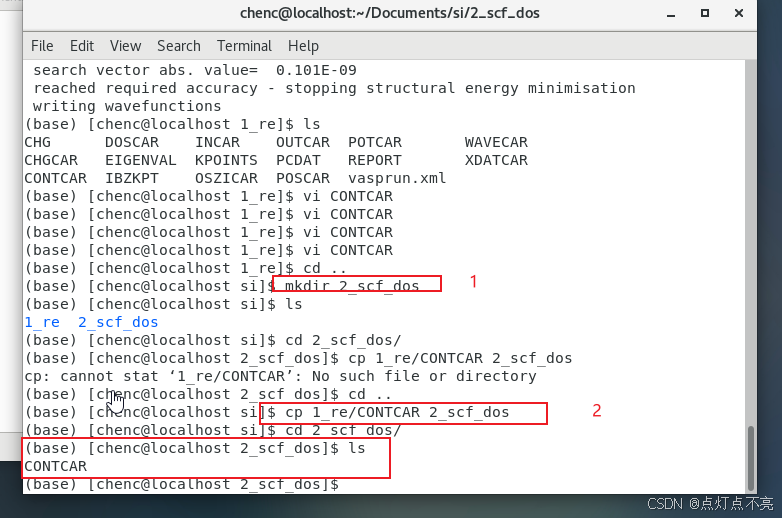
1、创建第二个文件2_scf_dos
2、将1_re中的CONTCAR复制到2_scf_dos中
2.2.2、INCAR
打开vaspkit,依旧选择生成输入文件
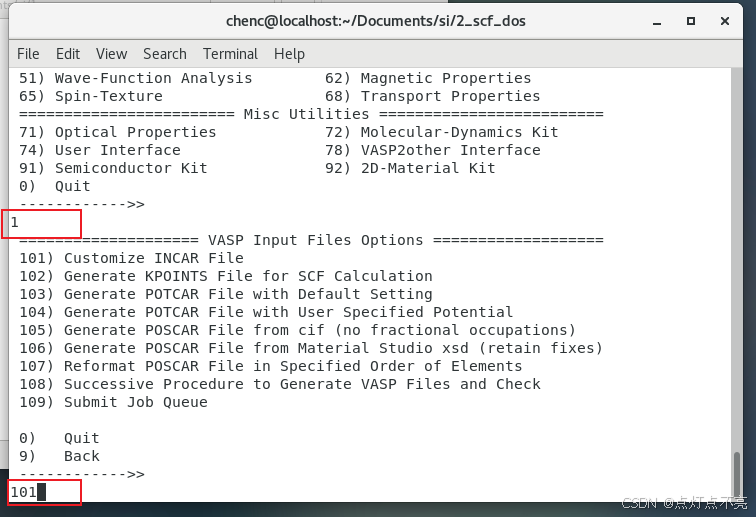
注意这里不需要再做任何的结构优化,选择静态计算
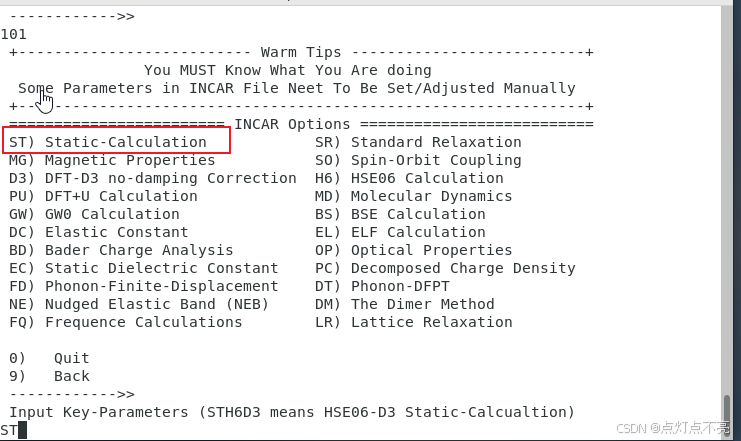
2.2.3、KPOINTS、POTCAR
继续使用vaspkit生成KPOINTS和POTCAR
报错信息
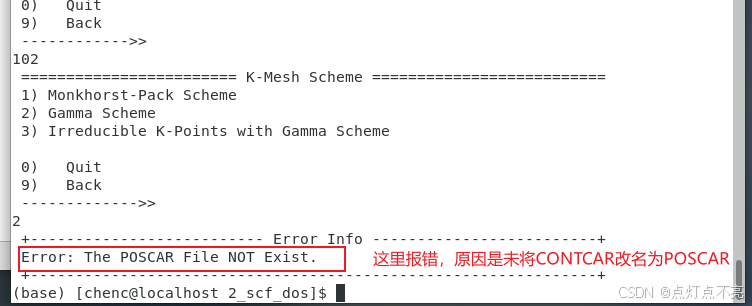
使用mv命令修改文件名
mv CONTCAR POSCAR
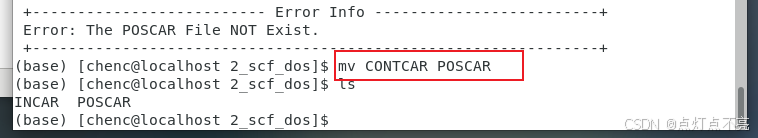
改名后使用vaspkit生成KPOINTS和POTCAR
2.2.4、TDOS.dat文件
计算完后使用vaspkit获得态密度

阅读并写入态密度数据到TDOS.dat文件

2.2.5、绘图
将TDOS.dat用orginpro工具打开
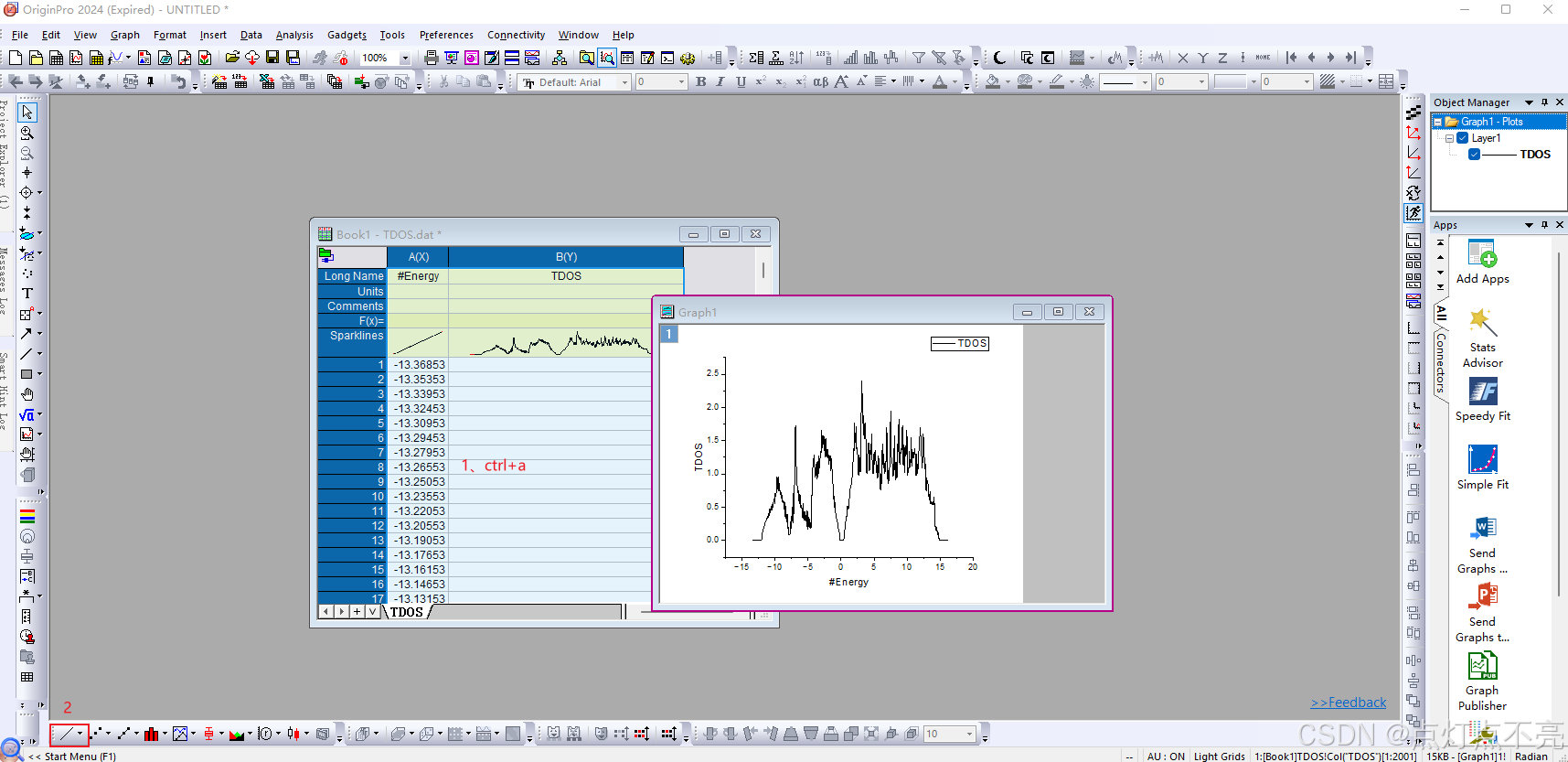
与官网的图做对比:
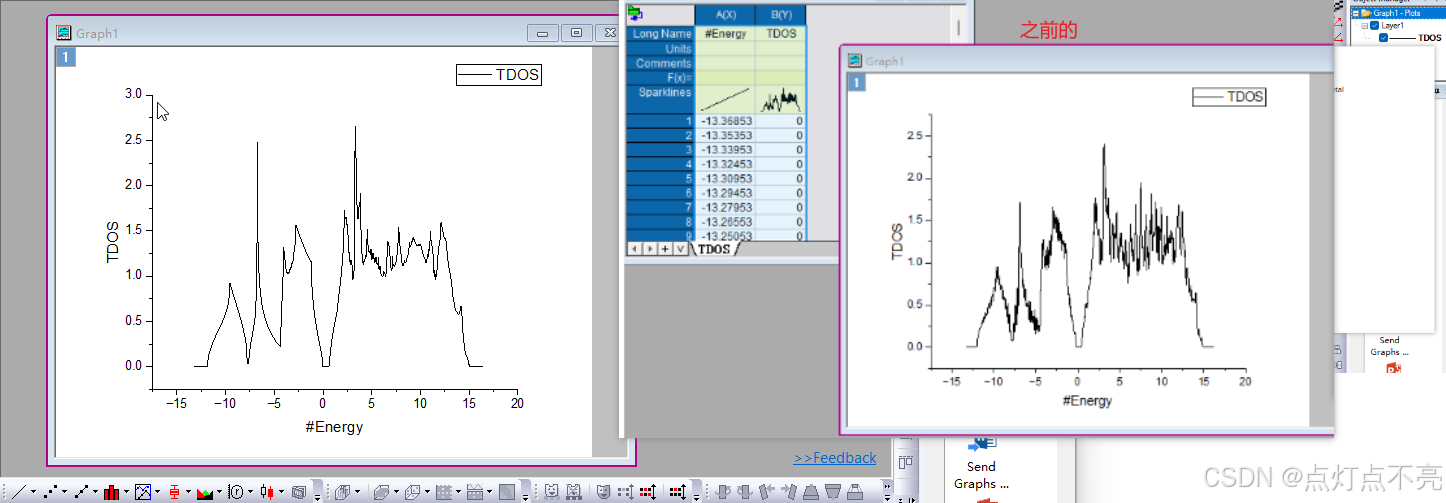
注意由于我的INCAR没有将ISMEAR 设置为 -5 ,所以图像看起来不平滑且毛刺很多
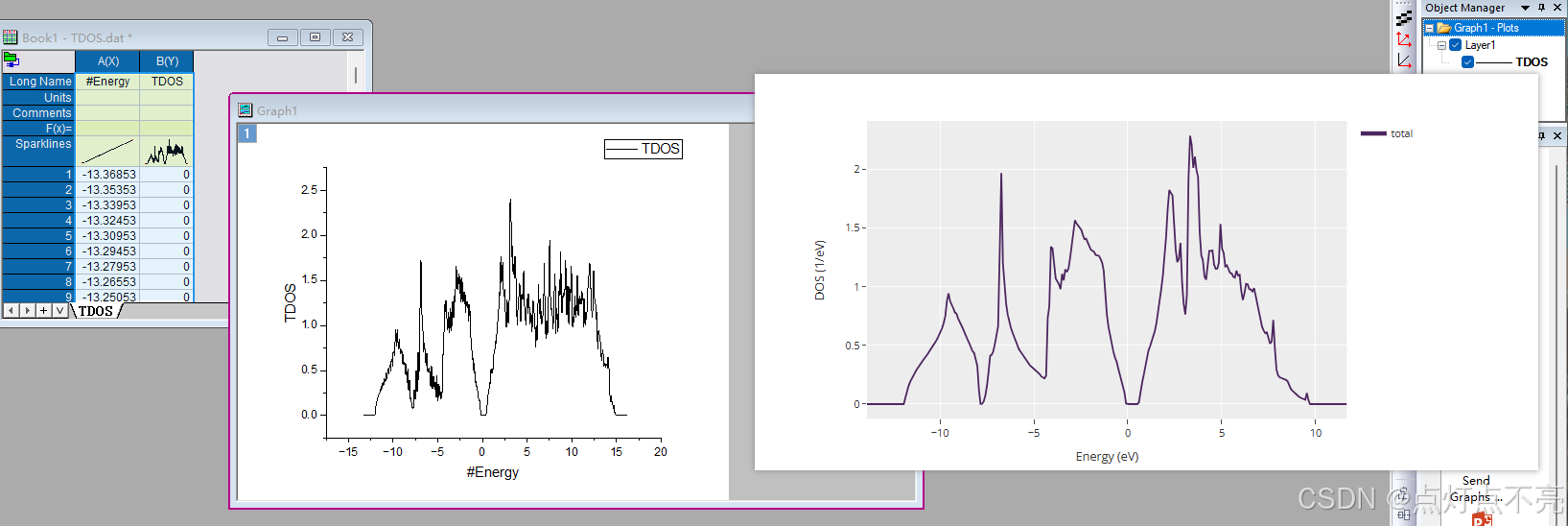
修改INCAR 中的ISMEAR参数为-5后重新提交计算后才获取态密度文件
将TDOS.dat用orginpro打开并于之前的做对比
针对四面体结构计算使图像毛刺少了很多,图像变平滑了
三、计算能带
3.1、官网解读
3.1.1、输入文件
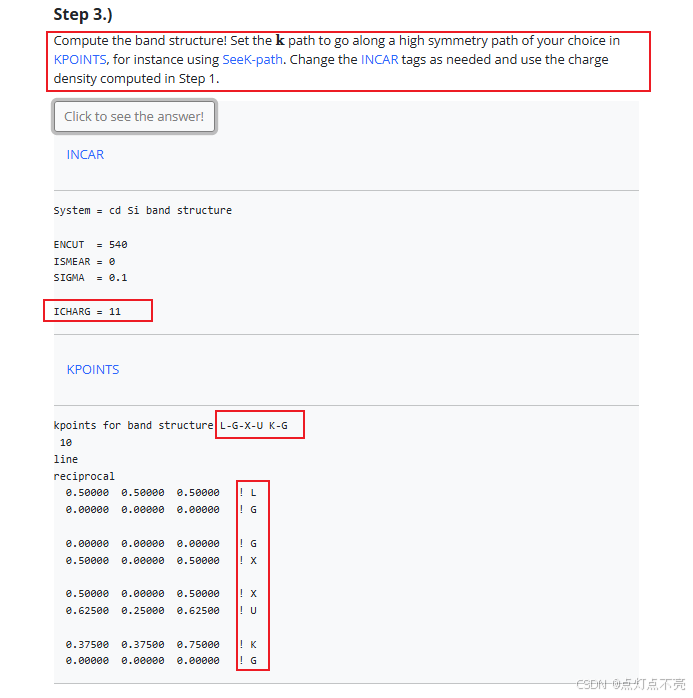
1、设置高对称点K路径,把布里渊区分割成多条路径,然后沿路径均匀撒点。官网给出的路径为L-GAMMA;GAMMA-X;X-U;K-GAMMA。
2、ICHARG决定VASP如何构建初始电荷密度。ICHARG设置为11:从CHGCAR读取给定电荷密度的本征值(用于能带结构图)或态密度(DOS)。自洽CHGCAR文件必须通过完全自洽的计算预先确定,该计算使用跨越整个布里渊区的k点网格。
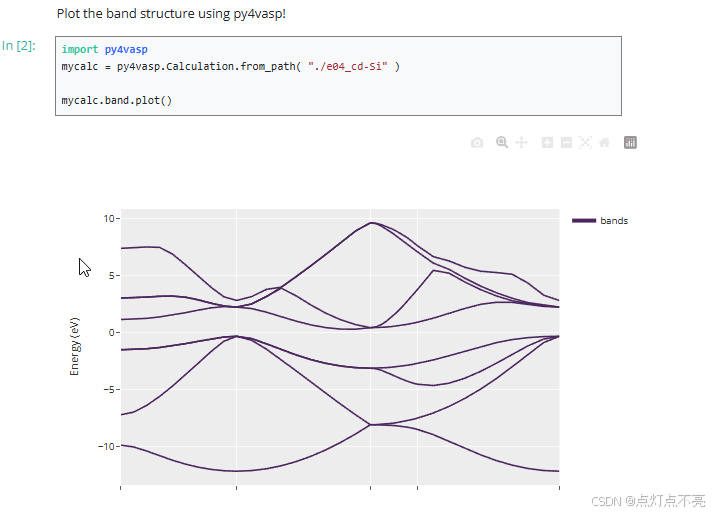
3.1.2、作图
3.2、自行计算
3.2.1、POSCAR
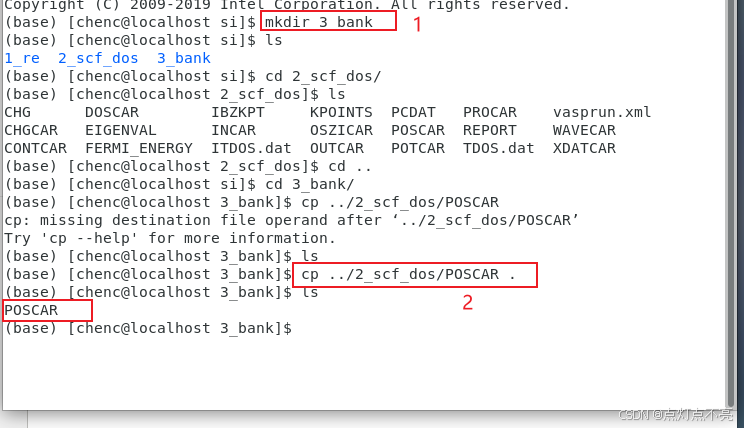
1、创建第三个文件3_bank
mkdir 3_bank
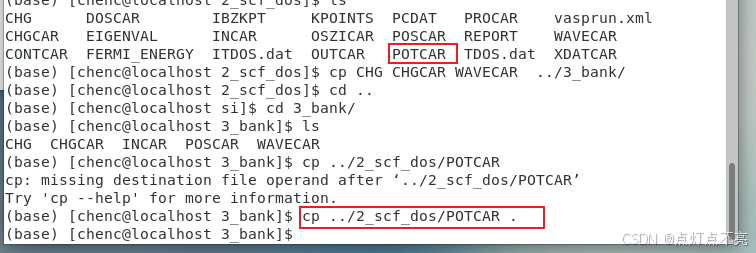
2、将第二个文件的结构文件(POSCAR)拷贝到第三个文件里
cp ../2_scf_dos POSCAR .
3.2.2、INCAR
我们的INCAR与官网的区别在于:
1、官网设置:ISMEAR = 0;ICHARG = 11;即不读取波函数文件只读取电荷密度文件CHGCAR,所以只用到了第二步计算生成的CHGCAR文件;
2、我们设置:ISMEAR = 1;即从现有的 WAVECAR 文件中读取波函数,并且默认地从头计算电荷密度。如果没有 WAVECAR 文件,VASP 会随机初始化电荷密度并进行自洽计算。
3、我们将第二步计算生成的CHG、CHGCAR、WAVECAR文件拷贝过来,这样是否使用ICHARG都能获取到电荷密度。
4、CHGCAR 是标准的电荷密度文件,包含了网格上的电子密度分布。CHG 文件是较旧的版本,但其内容和 CHGCAR 基本相同。
使用vaspkit生成INCAR
再重新静态计算一遍,求得的本征值就是能带(选择ST)
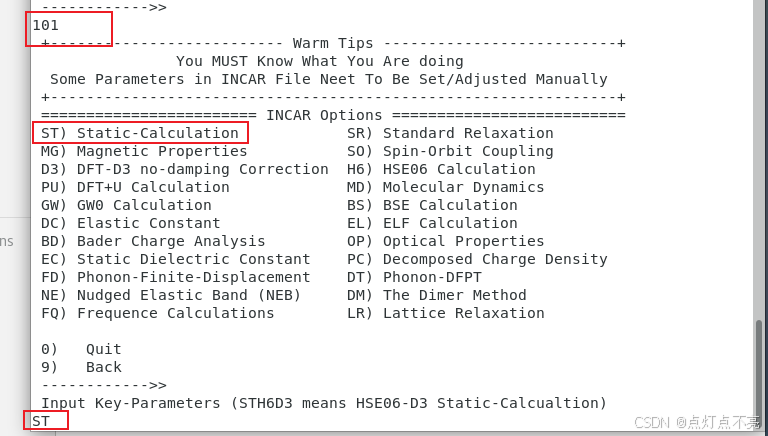
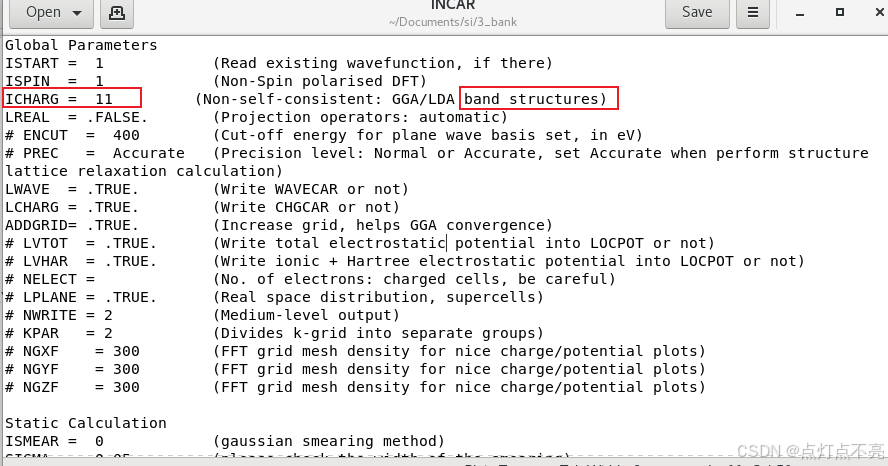
注意INCAR这里ICHARG被注释掉了
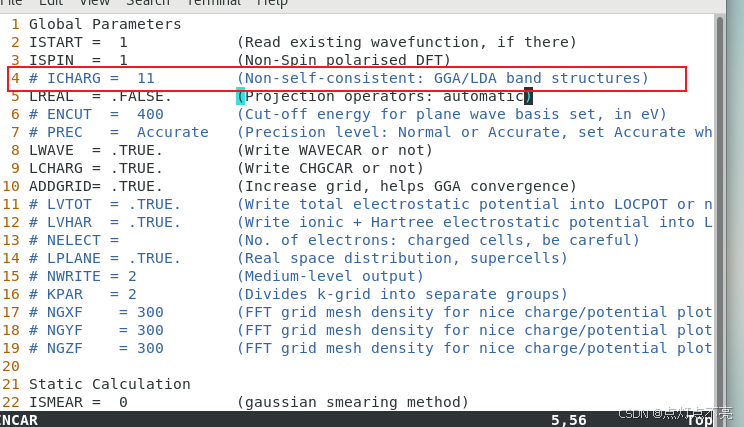
官方例程则表示ICHARG要设置为11
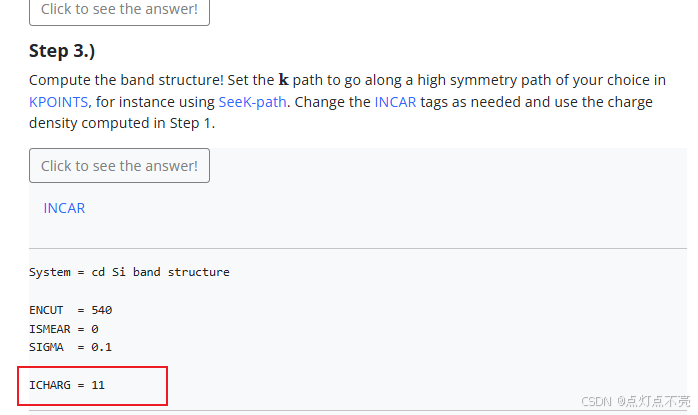
查询VASP手册ICHARG说明
ICHARG决定VASP如何构建初始电荷密度
ICHARG设置为11:从CHGCAR读取给定电荷密度的本征值(用于能带结构图)或态密度(DOS)。自洽CHGCAR文件必须通过完全自洽的计算预先确定,该计算使用跨越整个布里渊区的k点网格。
所以这里需要第二步自洽计算生成的CHGCAR

将需要的文件拷贝过来(这里为什么拷贝三个文件上面已有解释)
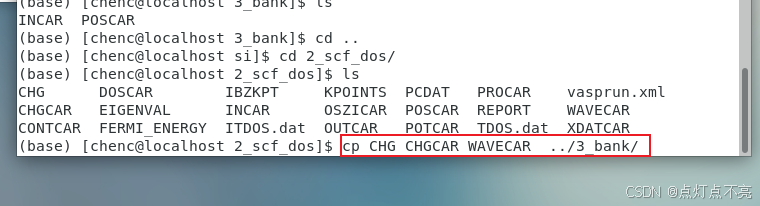
再将POTCAR赝势文件复制过来
在INCAR中将ICHARG设置为11(这里是否设置都可以,上面已有解释)
3.2.3、KPOINTS、POTCAR
使用vaspkit生成KPOINTS
这里选择能带结构的工具包,这样设置为自动选择高对称点的路径
这里提示生成的是KPATH.in文件,而我们提交计算需要KPOINTS
拷贝一份KPATH.in并重新命名为KPOINTS
所需输入文件准备好后提交计算
跑完之后使用vaspkit生成能带
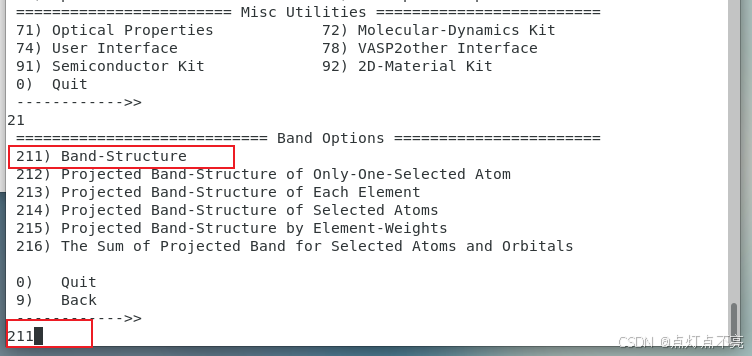
3.2.4、能带文件
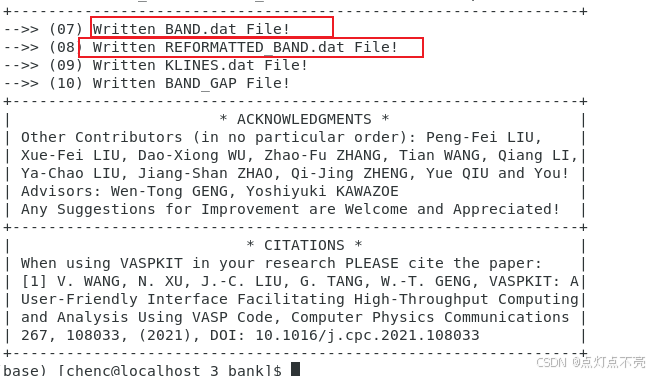
两个文件对比发现REFORMATTED_BAND.dat更利于绘图
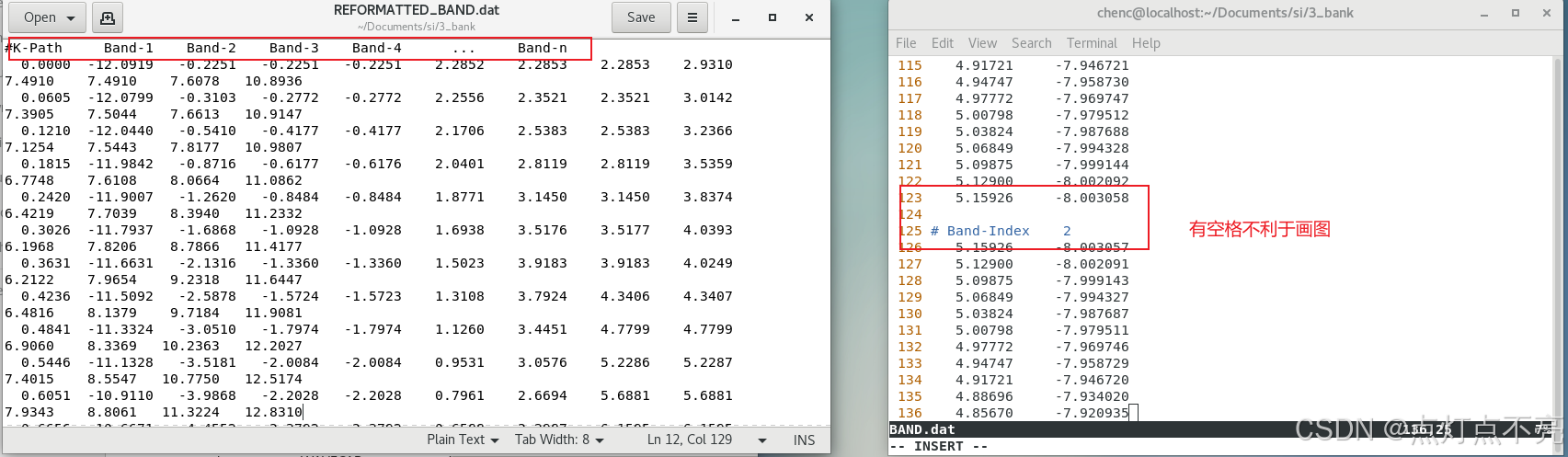
3.2.5、绘图
将REFORMATTED_BAND.dat用originpro打开
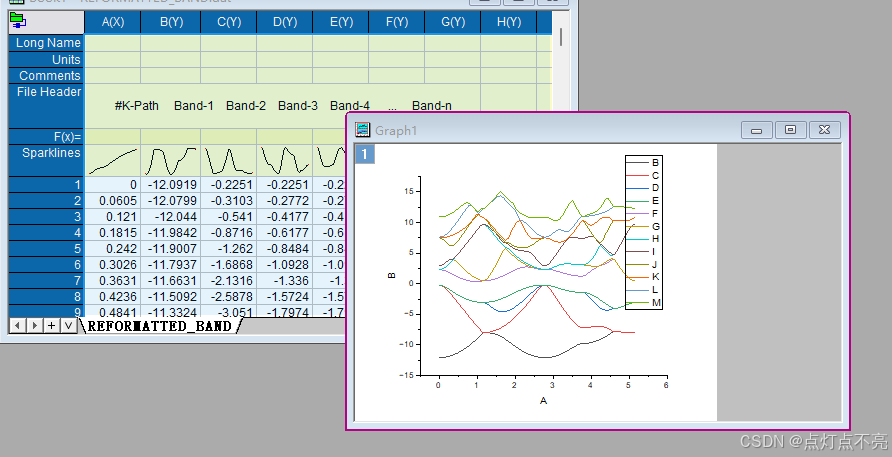
将文件头复制到longname里
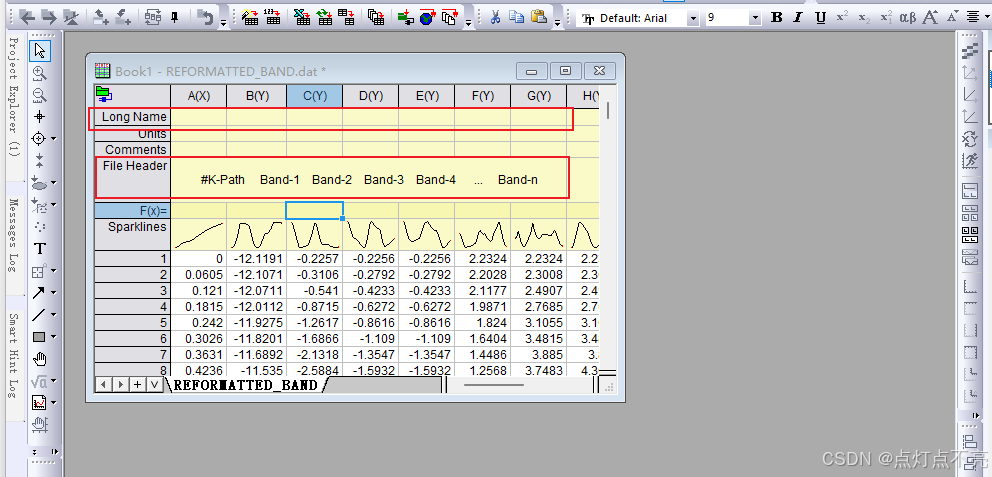
删掉多余的部分
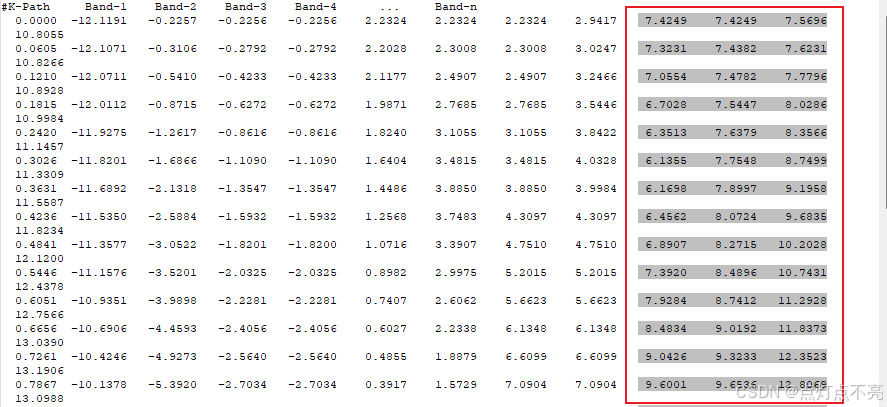
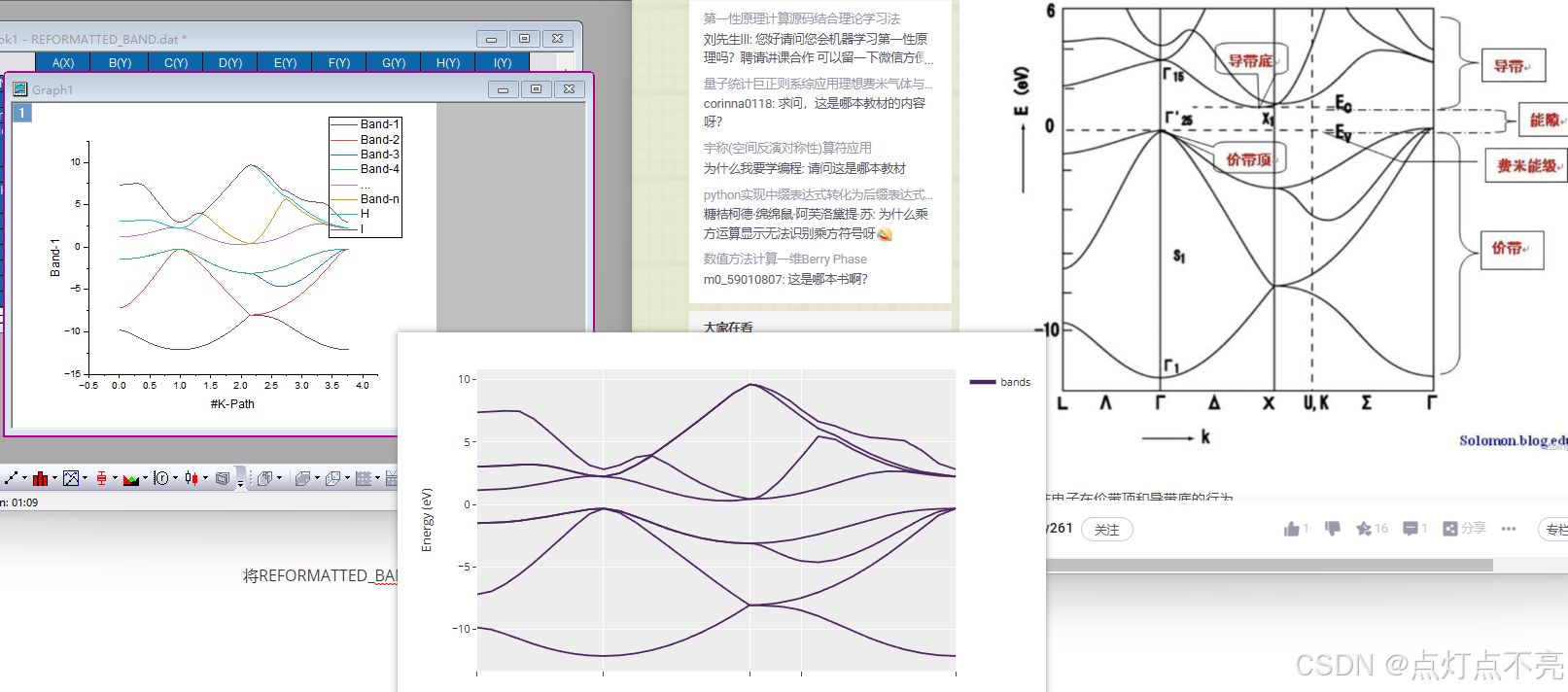
与官方例程做对比,发现是镜像对称
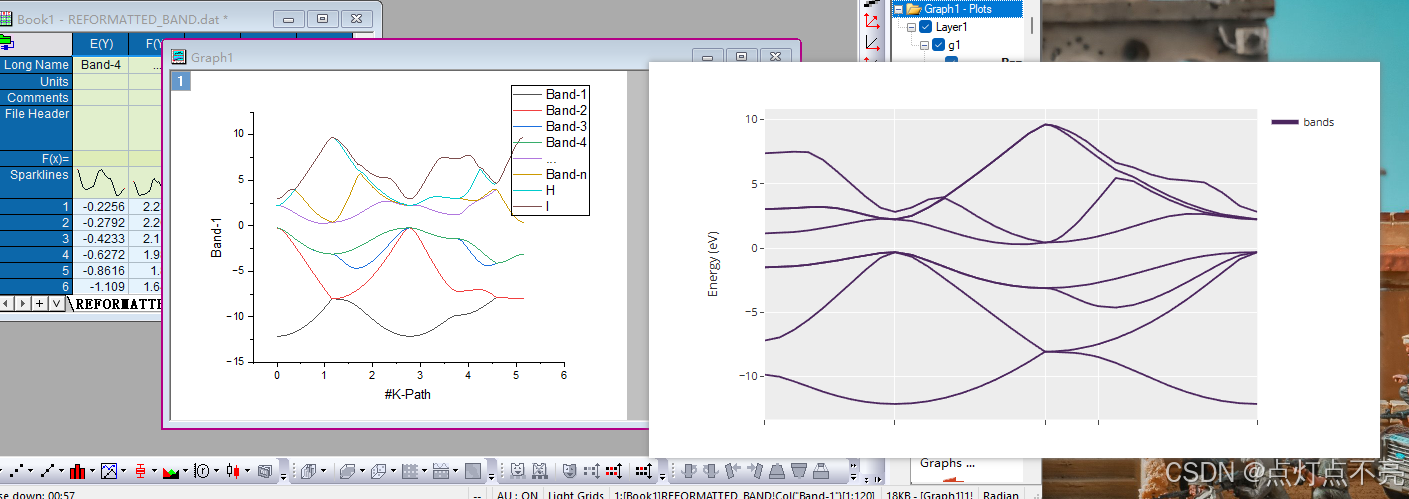
3.2.6、修改KPOINTS路径
查询硅的能带图获取正确路径(官网的例程也给出了)
发现其路径为L->GAMMA; GAMMA->X; X->U;; K->GAMMA;
注意:U和K共享一个点,故UK不作为一条路径

修改后的KPOINTS路径
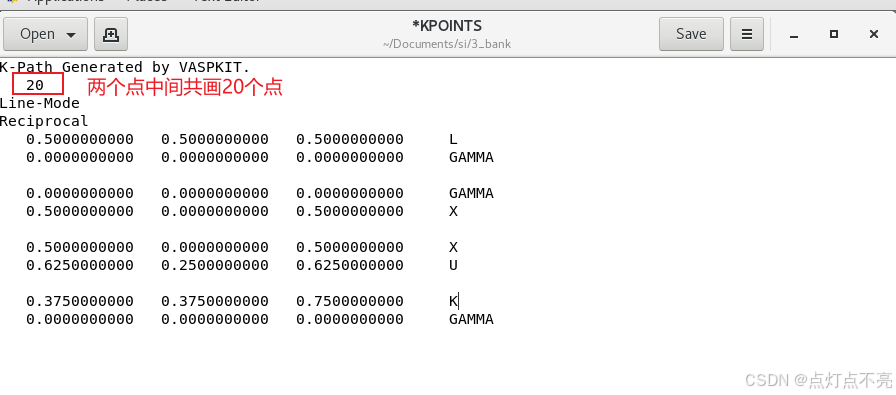
删掉之前生成的KPATH.in并重新提交计算
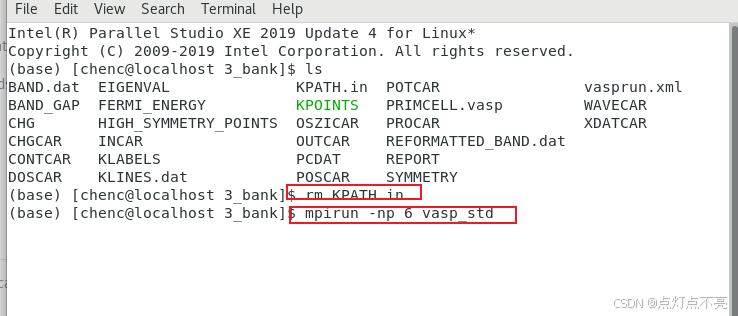
再次使用vaspkit生成能带相关的输出文件
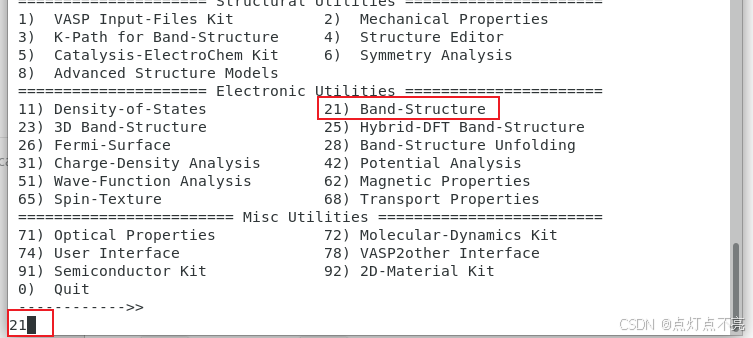
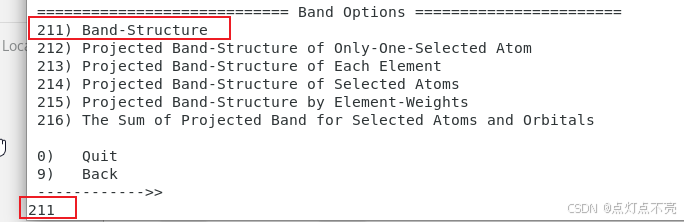
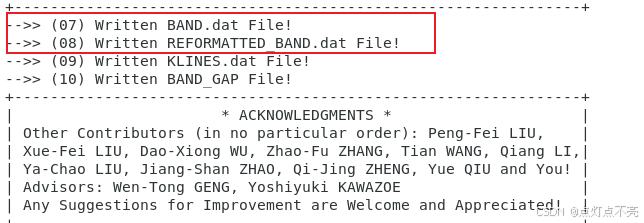
3.2.7、再次绘图
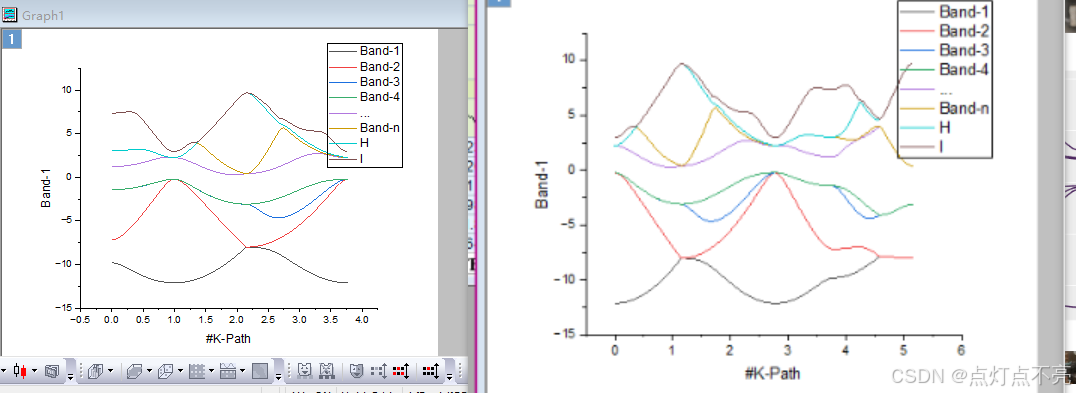
将REFORMATTED_BAND.dat用originpro打开
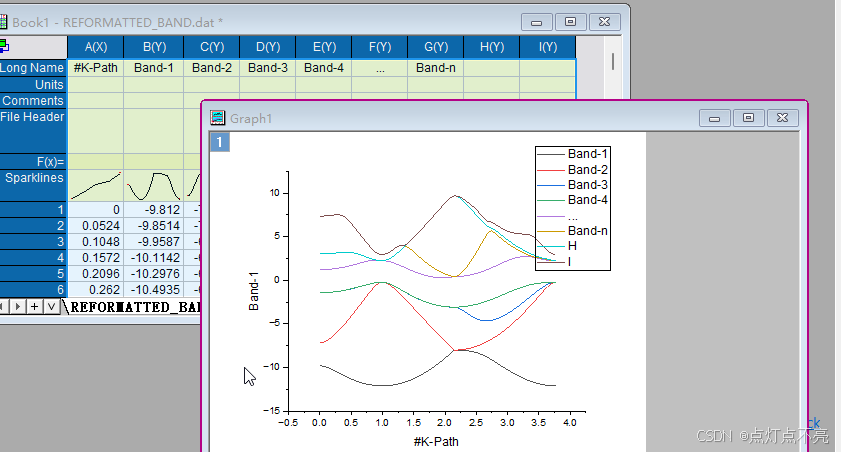
与官网和csdn对比
与修改路径前对比

















 882
882

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









