







reading notes of《Artificial Intelligence in Drug Design》
文章目录
- 1.Introduction
- 2.Tools for De Novo Design
- 3.Adoption of AI and Generative Models in Drug Discovery
- 4.From Early to Present Days
- 5.Use of Generative Models : Distribution Learning Versus Focused Learning
- 6.Applications in Drug Discovery
- 7.REINVENT : Uses Generative Models
- 8.De Novo Design of Libraries
- 9.Challenges in the Application of AI and Future Development
1.Introduction
- While the prediction of properties isn’t an exclusive AI/deep learning domain, it does benefit from the architectures of deep neural networks (DNN).
- AI is naturally seen as a means to automate activities in a flexible and adaptive way. The more routine the activity is, the easier it is to automate it.
2.Tools for De Novo Design
- The goal of de novo design is to identify novel active compounds that can simultaneously satisfy a combination of essential optimization goals such as activity on primary target, selectivity against off-targets, physicochemical and ADMET properties.
- Probably the biggest success of this period stems from the idea to couple fragment libraries with genetic algorithms (GA).
- The general GA operate on data represented as binary strings, which encode candidate solutions in the search space, and perturbs these strings thus mimicking the events of natural recombination and mutation in DNA.
- The majority of these methods however depend on a common denominator: a library of building blocks—predefined fragments. This has a significant impact and biases the chemical space that will be generated.
- The search space is clearly discontinuous since it relies on fragments as the building blocks which makes the gradient driven navigation less efficient.
- One evident advantage being the fact that fragment libraries offer only fragment resolution in contrast to the atomic resolution of generation of novel ideas that the generative models provide.
- It is much more efficient to also pair up the generative models with a proper search algorithm (SA), ideally a reinforcement learning (RL).
- Such tool should be equally effective and capable of finding optimal results for both exploration and exploitation scenarios:
- Exploitation—when we are aiming to optimize a promising idea and we do not want to move too far away in the search space in terms of core scaffold similarity. This mode is relevant when optimizing properties of a given lead.
- Exploration—when finding good ideas is desirable but it is even more important to have ideas that are diverse and belong to different regions of the chemical space. This is particularly needed when we aim for scaffold hopping in order to escape patents or unfavorable solution space.
3.Adoption of AI and Generative Models in Drug Discovery
- In addition to building the molecules atom by atom or token by token researchers have explored the possibility to use fragment intermediates as building blocks.
- There has been also a recent effort in developing an alternative string format such as SELFIES. SELFIES which stands for Self-referencing Embedded Strings is aiming to address the fact that a large portion of SMILES strings do not actually correspond to any valid molecules while every SELFIES string corresponds to a valid molecule.
4.From Early to Present Days
- These pioneering works were still suffering from various shortcomings such as high percentage of invalid structures, various performance issues, significant bias and lack of ability to navigate efficiently the chemical space.
- Some of these works are available as open source: GENTRL by Insilico Medicine, MSO by Bayer and REINVENT by AstraZeneca.
- The fashion in which the graphs are encoded as an input to the networks resembles the way circular fingerprints (ECFPs) are built. The relevant atom features are initially encoded into each node and then this information is iteratively passed to the neighboring nodes, by normally following one of the Message Passing Neural Networks (MPNN) framework’s methods.
- However, the applicability for generative tasks still appears to be limited due to the memory demanding nature of the graphs especially when compared to SMILES.
5.Use of Generative Models : Distribution Learning Versus Focused Learning
-
Regarding the usability of the generative models we observe two main trends for de novo design: distribution-learning and goal- directed generation
- Distribution-learning efforts are mostly focused on generating ideas that resemble a particular set of molecules. This is commonly achieved in three steps: subjecting the generative models to transfer learning (TL), subsequently sampling the resulting model thus generating a fast body of data and finally evaluating the resulting data. It is also clearly, less efficient and much more computationally demanding method in comparison to the goal-directed generation.
- The alternative approach of goal-directed generation is conducted by using some form of a search algorithm while aiming to find molecules that satisfy the given objective, without having to sample the entire search space. This is conducted in three steps: sampling, scoring and learning.
-
From user’s perspective however it is much more important to understand how to use the generative models in order to achieve either exploration or exploitation of the chemical space. For exploitation, users define an area of interest and focus on generating compounds that share similar structural features. In contrast, the exploration mode enables them to obtain compounds that share less structural similarity but still satisfy other desired features.
6.Applications in Drug Discovery
- The earliest study where AI generated ideas were successfully synthesized and tested, appeared in 2018 by the Schneider group. Their model use transfer learning and LSTM cells generate 49 structures. Finally four of them demonstrate considerable potency and diverse selectivity profiles on PXRs and PPARs.
- A recent work that received widespread attention by Zhavoronkov et al. demonstrated the use of a deep generative model, named as generative tensorial reinforcement learning (GENTRL), to discover potent novel inhibitors of discoidin domain receptor 1 (DDR1), a kinase target implicated in fibrosis and other diseases. Notably the design was complete after only 21 days.
- Pickett et al. from GSK have presented in 2019 a deep generative model that is capable of translating a Reduced Graph to SMILES strings.
- In 2019 Winter et al. from Bayer introduced a new model that was also inspired from advances in neural machine translation. It translates between two semantically equivalent but syntactically different representations of molecular structures, compressing the meaningful information both representations have in common in a low-dimensional representation vector.
- The PSO optimisation (Particle Swarm Optimization) tool appeared as a component of gru ̈nifai an interactive in silico compound optimization platform that was released in 2020 by Winter et al…
- The same encoder/decoder NN was used as a plug-in in QMO, a generic query-based molecule optimization framework that exploits latent embeddings by Hoffman et al. from IBM Research. They introduce a novel query-based search method to achieve efficient optimisation with discrete molecules.
- Me ́ndez-Lucio et al. also from Bayer used a similar encoder/decoder NN framework coupled with a conditional generative adversarial network (GAN) to generate molecules conditioned on transcriptomic data.
- Grebner et al. from Sanofi-Aventis evaluated in-depth the influence of different chemical spaces as input training sets and different scoring functions and their combinations during training of a generative model with focus on application on real-life project support workflows.
- The model was trained with three different datasets with the idea that “ChEMBL” represents a diverse set, “Enamine” a synthetically accessible and “Sanofi” an industrial quality one.
- (1) ChEMBL24 (~1.45 million molecules),
- (2) a subset of Enamine REAL space (~5.36 million molecules),
- (3) a subset of Sanofi compound collection (~3.37 million molecules)
- A diverse set of scoring functions was considered: 2D similarity based, 3D similarity based using ROCS from OpenEye, 2D QSAR model, aggregate score of 2D similarity, 3D similarity, and QSAR as a sum and as a product.
- 2D similarity-based scoring favors generation of novel structures but around known chemotypes (exploitation) and it thus offers an interesting alternative to transfer learning strategies.
- Excluding particular substructures from the training set input shows only a minor effect confirming the ability of the generative NN to generalize. For example, the model learns to generate structures with rings of a new size that were not present in the training input.
- Due to stochastic nature of the generative NN, multiple runs of the same design (identical prior, scoring function, number of optimisation steps, etc.) can yield results from different regions of the chemical space. Higher variability was found with more complex scoring functions.
- Diversity and high quality of the training dataset is more important for molecular design than dataset size.
- Using 3D shape similarity or QSAR scoring functions produces significantly different design ensembles, as these scoring functions allow for sampling of a much broader region of the chemical space and are more suitable for lead generation.
- For generating analogue ideas, in a lead optimization scenario for example, 2D similarity scoring appears to be more suitable although it can produce more conservative results.
- The model was trained with three different datasets with the idea that “ChEMBL” represents a diverse set, “Enamine” a synthetically accessible and “Sanofi” an industrial quality one.
- One more study by Amabilino et al. from GSK in 2020 is based on an adaptation of REINVENT as the generative NN where they investigate best practices to create a well-performing transfer learning application and provide practical guidelines for optimal use.
- The performance of the generative model is measured by six metrics, following the GuacaMol benchmark: the percent- age of valid generated SMILES, the percentage of unique SMILES, the percentage of “novel” molecules (i.e. not already present in the transfer learning data set), the percentage of both novel and unique molecules, the Frechet ChemNet score, and the Kull- backLeibler score.
7.REINVENT : Uses Generative Models
- REINVENT is a de novo design tool which is developed at Astra- Zeneca. It is essentially a Recurrent Neural Network (RNN) architecture with a long short- term memory (LSTM) cell. It is trained on data derived from ChEMBL in SMILES format. Therefore, the model learns the SMILES syntax and is capable of producing correct strings with validity of above 99%.
- In REINVENT searching is achieved by subjecting the generative model to a Reinforcement Learning (RL) scenario with policy iteration while aiming to satisfy a set of requirements that presumably define the desired compounds.
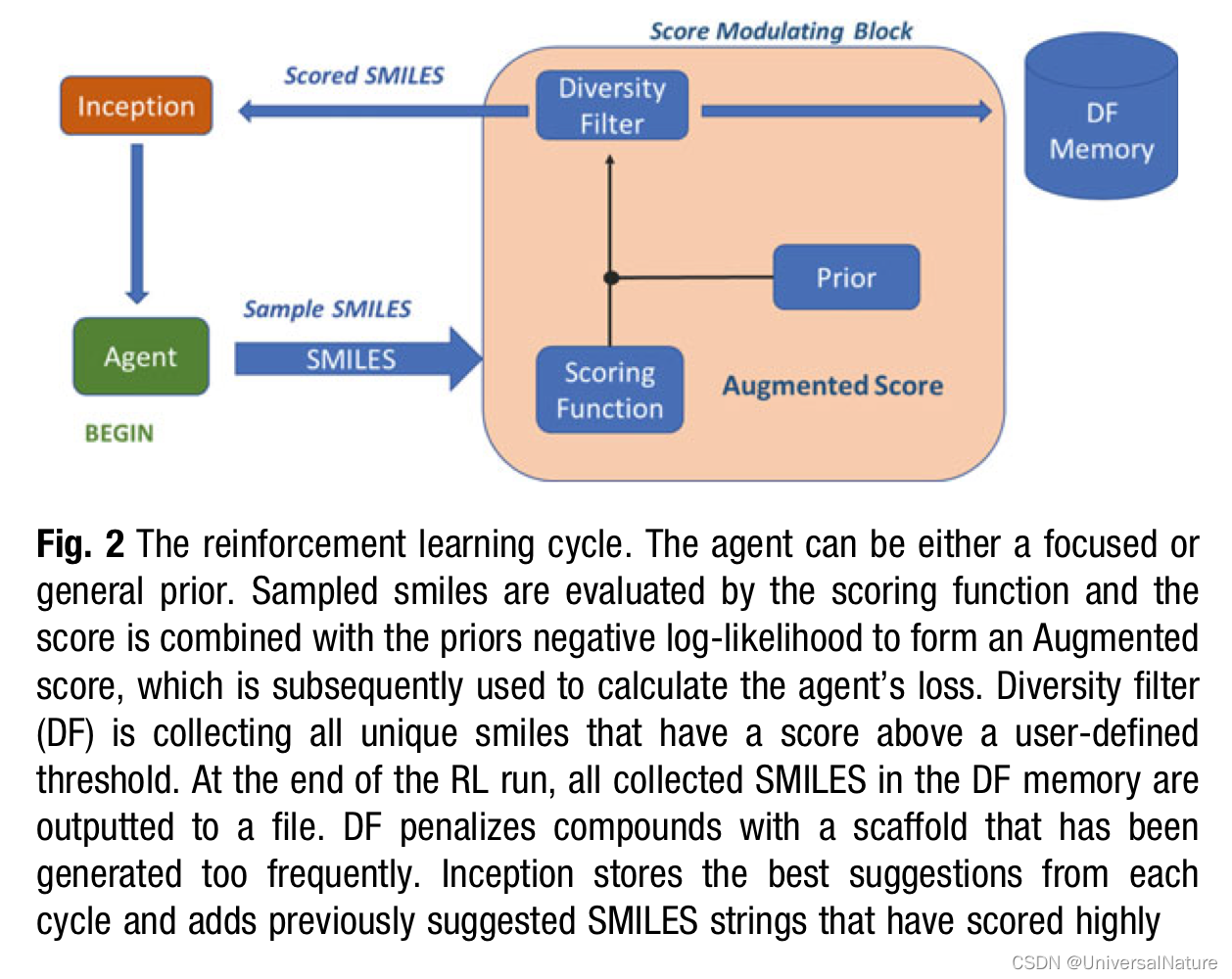
-
Maximizing the outcome of a given scoring function may sometimes lead to get repeatedly stuck in a narrow space of solutions. This would in turn lead to a mode collapse of the generative model. In order to avoid such scenario REINVENT uses diversity filters (DF).
-
DF prevents from gaining reward when generating the same or similar compounds recurrently. This is achieved by memorizing the generated compounds.
-
Conducting a successful RL run can sometimes be challenging especially if the SF is composed by very strict components or by components that are often orthogonal. This would lead to longer learning times and reaching the state of productivity much later, thus generating a lower yield of compounds with a score that satisfies us. To help the learning process we may resort to prefocusing the generative model via TL. The focused model can be subsequently used as a starting point in the RL.
-
An alternative to prefocusing the generative model is to use “inception.” The inception feature in REINVENT is a modified version of experience replay.
-
A notable feature of REINVENT is the ability to combine together a variety of factors into a single scoring function. However, bringing multiple components together within a single generation run can hold some challenges. Therefore it is important to find the right balance and to include the most relevant factors so that the learning process is conducted at optimal speed and the generated compounds are scored reliably.
8.De Novo Design of Libraries
- A frequent use case in drug design projects is to explore properties of compounds that share same scaffold. A particular advantage of such libraries of similar compounds is usually the ease of synthesis.
- While retaining the scaffold is perfectly feasible for the core REINVENT implementation, it is more challenging to enforce certain constraints so that the scaffold is guaranteed to be modified only at a specific point or at several points at a time. Therefore an alternative implementation aiming at design of libraries of small molecules was developed.
- The purpose of the RF being to enable users to provide additional requirements for the generated compounds. These constraints are in the form of retrosynthetic reaction SMIRKS chosen by the user.
9.Challenges in the Application of AI and Future Development
- The importance of data size and quality is well established and understood and in the context of de novo molecular generation has been illustrated by some of the examples in this review.
- Further enhancement of data size and diversity can be achieved by data augmentation techniques, as in the generation of multiple SMILES representations for a molecule by ArusPous et al. which they found beneficial for RNN based molecular generation.
- Availability of high-quality data is also important. A large amount of public data is now accessible from databases such as PubChem and ChEMBL. There is however, a significant amount of proprietary data that is not publicly available and is only accessible within individual biopharmaceutical companies.
- For data driven learning, the NN architecture together with the representation type of a molecule are of equal importance to data as they define the inductive bias of the model. It has been already shown from examples in this review how different architectures (e.g., RNN, VAE, graph NN) and different molecule representations (e.g., SMILES, SELFIES, graph, image) can affect generative performance.
- Currently a major limitation of the vast majority of deep generative models is that they can only represent and process two-dimensional information for molecules and thus cannot distinguish between stereoisomers that is molecules with identical 2D but distinct 3D orientation which can have completely different biological activity profiles. One of the natural approaches would be to extend the convolutional neural network (CNN) architecture in order to handle three-dimensional data as opposed to the standard 2D CNN that handles flat images.
- A different approach in the context of RL would be to incorporate 3D information not in the representation stage but by using a 3D enabled reward function.
- Using docking has the added advantage that it is physics-based while predictive models rely solely on activity data. Use of the latter category of models has attracted criticism in the context of molecular generation, as generative performance is dependent on the availability and quality of training data and can become poor outside the applicability domain of the predictive model.
- Optimization for Medicinal Chemistry and for Drug Discovery in general is inherently multi-objective where several objectives need to be balanced and it is not unusual that a subset of those show, in pairs, negative correlation thus improving one can deteriorate the other and with the added complication that these relationships can be highly nonlinear.
- There is no doubt that this is a challenge of critical importance for Drug Discovery and far from acquiring a global solution. There are two main components for this challenge:
- Computational. A complete solution in the general case can be computationally expensive or even impossible because of nonlinearity, optimization spaces that are multidimensional or discrete, etc.
- Conceptual. In many cases, not all the objectives are known that need to be combined together to achieve an optimization goal. In other cases, even when an objective is defined, it cannot be encoded to a suitable mathematical form that can be utilized by an optimization algorithm. Additionally, optimzation in Drug Discovery is iterational in nature where new data are collected in every iteration adding up new knowledge that needs to be incorporated into the optimisation process. Those issues make human participance indispensable but also point to a powerful approach that combines AI with human interaction, known under the general term of Human-in-the-loop (HITL).
- Of central importance to small molecule Drug Discovery is the synthetic accessibility of any proposed molecules, even the best ideas are of little use if they cannot be realized in the synthetic lab with sufficient purity and quantity and in a reasonable time and cost. This is a well-known and described issue for generative models, a recent discussion can be followed in the work by Gao and Coley.
- A different approach by Bush et al. from GSK introduced a new benchmark aligned with human expert intuition as a threefold framework that tests: (1) human inclusion, the ability of AI to reproduce human ideas, (2) human imitation, where medicinal chemists rate AI ideas, and (3) Legacy projects, with the aim of testing the ability of AI to replicate molecules in legacy drug discovery projects, given a single seed molecule from the series.
- Moving away from the technical side that we have described so far, there remains a major challenge for fruitful integration of AI into Drug Discovery which is cultural: no progress can be made if the scientific community is not willing to apply any of these methods or if there is lack of trust or support from the business in adopting them.
















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











