







reading notes of《Artificial Intelligence in Drug Design》
文章目录
- 1.Introduction
- 2.Materials
- 3.Methods
- 3.1.Generation of Chemical Structures
- 3.2.Training Generative Networks
- 3.3.Reward Functions for AI-Based De Novo Design
- 3.3.1.Physicochemical and ADME Properties
- 3.3.2.2D-Similarity Approaches
- 3.3.3. 3D-Similarity Approaches
- 3.3.4. Machine Learning Models for Scoring
- 3.3.5.Validation of Machine-Learning Models
- 3.3.6.Multidimensional Compound Optimization
- 3.3.7.Reward Functions in Latent Chemical Space
- 3.3.8.3D-Docking Based Reward Functions
- 3.4.Chemical Feasibility
- 3.5.Integration of Scoring Functions in Internal Workflows
- 3.6.Compound Selection and Filtering
- 3.7.A Practical Example: AI-Design at Work
- 4.Summary
1.Introduction
- De novo design is not restricted by availability of building blocks or a given set of chemical reactions and thus allows a potentially broader sampling of drug-like chemical space. However, synthesis of de novo designed molecules has often proven to be challenging.
2.Materials
3.Methods
3.1.Generation of Chemical Structures
- For generative networks, SMILES strings have emerged as one of the most simple, yet productive approaches of representing and generating molecules.
- Methods using fragment representations are used for a long time, while Graph-based methods have emerged more recently.
- First methods using 3D-based molecular representations are now becoming available as well.
3.1.1.SMILES-Based Approaches
- Therefore, the employed training set should represent the interesting chemical space with sufficient examples. Typically, several hundred thousand molecules from databases containing drug-like molecules are used to train a robust molecule generator. Prior to the actual training process, training sets are typically filtered by unwanted chemical fragments and unfavorable physicochemical property ranges to focus more on the “drug-like” structural space.
- While for successful structure-based virtual screening, enumeration of possible tautomers and protomers is strongly suggested as part of the workflows to ensure that the state with optimal interaction possibilities is included, a consistent treatment based on for example neutral molecular states and canonical tautomers might be more useful for SMILES, 2D-fingerprint, and 2D-descriptor based approaches including similarity searching, machine-learning, and training database generation.
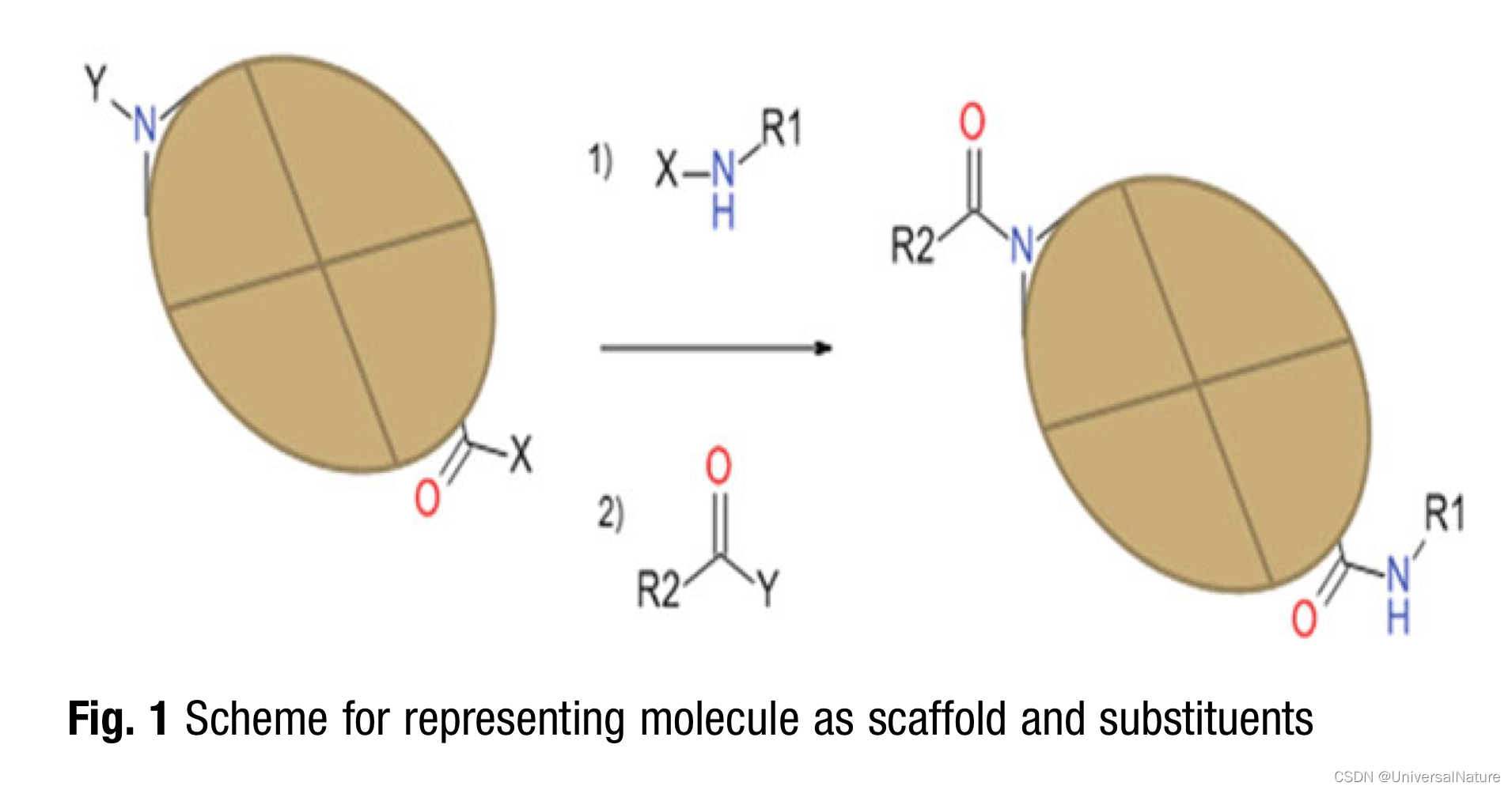
- During the optimization of lead structures for a particular activity, some parts of the molecules are often kept constant, if they contain essential pharmacophoric elements. Therefore, other methods use a definition of a central scaffold (for example the central brown box in Fig. 1) and maintain this scaffold during the optimization.
3.1.2.Autoencoder
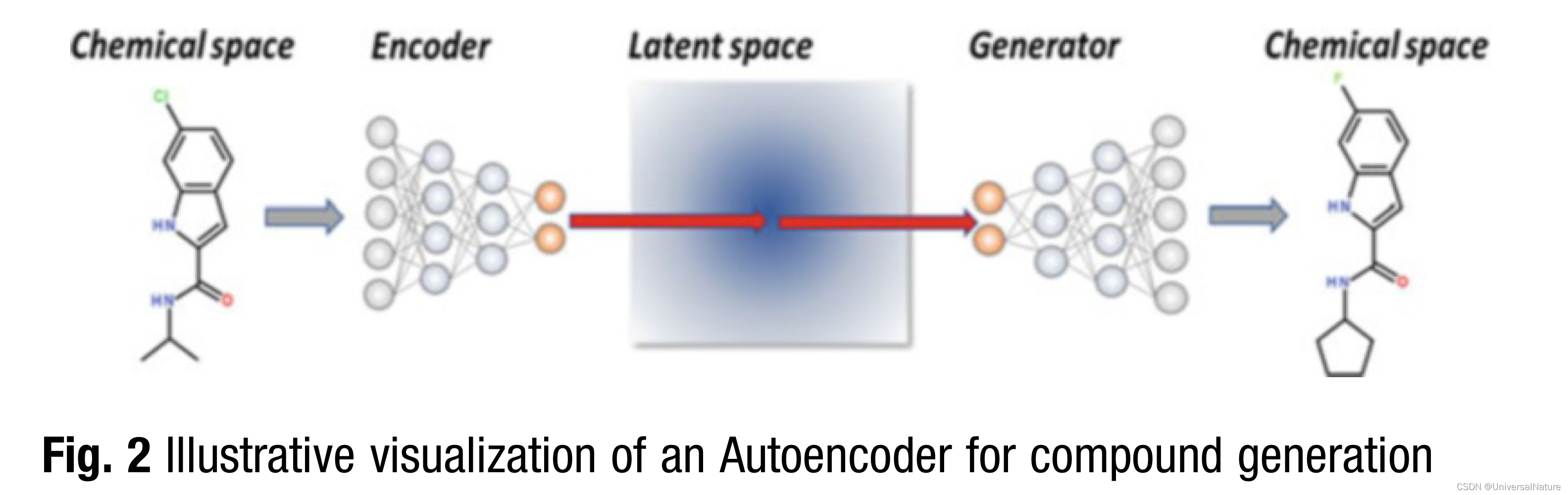
- A very promising method is the Junction Tree Variational Autoencoder (JT-VAE). For representing a molecule, it is first decomposed into a molecular graph and a junction tree.
3.1.3.Scaffold/Fragment-Based Approaches
- With the rise of deep neural networks and growing computational resources, fragment- or scaffold-based methods rose again. In this context, Stahl et al. developed a framework called DeepFMPO.
- Different rewarding schemes for AI-based design methods will be presented in Subheading 3.3.
3.1.4.Graph-Based Approaches
- One of the first approaches using graphs for generative models was introduced by Li et al…
- Another early approach is based on a variational autoencoder combined to a graph-representation of the molecules.
- Further on, Jensen used graph-based methods with genetic algorithms and Monte-Carlo tree search.
- Very recently, Mercado et al. introduced a graph-based design approach to generate optimized molecules called GraphINVENT.
- An advantage of graph-based methods is the encoding of chemical information in a more mathematical way which can be beneficial for neural networks. In contrast, the representation of a molecule is more abstract which makes understanding and interpretation of results harder.
3.1.5.3D-Based Approaches
- An implementation of a 3D-based approach was introduced by Fabritiis et al.
- Another approach is to use generative methods based on 2D-molecule representations, while the scoring and selection process for interesting molecules is based on 3D-methods like 3D-shape overlays.
3.2.Training Generative Networks
- In general, approaches to generate optimized compounds can be divided into three groups.
- Filter approach: A molecule generator samples molecules from the large chemical space, while a subsequent reward function acts as a filter and selects the most promising candidates.
- Biasing/Reward approach: A generator is coupled to an optimization/reward routine, which is biasing the generator toward the desired molecular properties. The bias is still unidirectional which means, the reward function is not influenced by the generator.
- Combined generation and reward approach: Here, generation and optimization/reward steps are directly combined, that is, a bidirectional communication between both methods.
3.2.1.Transfer Learning
- Early applications of SMILES-based approaches have used transfer learning to bias the neural networks which belongs to the first group of methods (“Filter approach”).
- One main limitation of transfer learning is the inherent requirement of an appropriate set of molecules to learn from.
- Successful applications for the design of novel molecules have been demonstrated.
3.2.2.Reinforcement Learning (RL)
- One of the main challenges is the definition of an appropriate, balanced scoring scheme. With an unbalanced scoring, the optimization can be trapped in local minima or driven into undesirable regions of chemical space.
- First successful applications of reinforcement learning for design were reported by Zhavoronkov et al.
3.2.3.Genetic Algorithms and Global Optimization
- Key to a successful application of a global optimization approach is a proper way of building and generating new molecules.
- In a recent study, Jensen explored a genetic algorithm with a graph-based representation of molecules, he also explored Monte-Carlo Tree Search as an optimization method. Both methods perform well in GuacaMol benchmark.
3.2.4.Generative Adversarial Networks
- The group of AspuruGuzik proposed a framework of GANs called ORGAN and applied it to chemistry and material design (called ORGANIC)
- Another method was proposed by Maziarka et al. who named their method Mol-CycleGAN, as it is based on the CycleGAN-algorithm
3.2.5.Autoencoders with Optimization Methods
- Winter et al. further proposed a method using a particle swarm optimization (PSO) together with the latent space to generate compounds of desired properties.
3.3.Reward Functions for AI-Based De Novo Design
- Hence, any useful function should rapidly and reliably identify virtual molecules showing favorable chemical motifs. Other approaches, which require significantly more computational power can only be applied at the end of such a workflow to further rank promising proposals.
- We perceive scoring as a two-step process by (a) including the most relevant and rapid functions directly into the generative design step and (b) using other scoring functions in a multidimensional fashion to select interesting molecules from the larger ensemble of virtual proposals by the AI-engine.
- Typical scoring functions capture molecular features based on similarity to previous lead molecules or statistical models summarizing previous project knowledge. Computational approaches for deriving these functions can be broadly grouped into the following categories:
- Physicochemical properties filters
- Ligand-based 2D similarity approaches
- Ligand-based 3D similarity approaches
- Machine-learning models from structure-activity data
- Structure-based 3D approaches
3.3.1.Physicochemical and ADME Properties
- Typical property filters for scoring use descriptors related to lipophilicity (e.g., logP, logD), surface area fractions (e.g., PSA), key functional group counts (e.g., number of donors or acceptors), measures of size and flexibility (molecular weight, number of rotatable bonds) recently reviewed by Mignani et al.
3.3.2.2D-Similarity Approaches
- 2D descriptors with a stronger scaffold hopping potential such as topological pharmacophores (e.g., CATS) provide opportunities to drive molecule generation toward chemically different scaffolds with a similar pharmacophoric pattern.
3.3.3. 3D-Similarity Approaches
- Nonobvious analogies between compound pairs beyond 2D chemical similarity can be obtained by 3D shape based technologies (e.g., ROCS).
3.3.4. Machine Learning Models for Scoring
- Experimental data from harmonized assay conditions at one laboratory or even at different research sites are essential to provide balanced training sets, while minimizing any associated experimental error.
3.3.5.Validation of Machine-Learning Models
- Therefore, the domain of applicability for a model should be investigated for each new molecule to be predicted.
- Valid confidence intervals for predictions as error estimations were provided by Bender et al. using the “Deep Confidence” framework.
- Many of our internal machine learning models apply a 2D fingerprint-based similarity threshold as applicability domain estimation.
- Moreover, the direct use of 2D-similarity to query molecules in combination with more advanced machine-learning models might intrinsically also limit the generation of novel virtual molecules to a region in chemical space relevant for lead optimization.
3.3.6.Multidimensional Compound Optimization
- A meaningful combination of different property prediction models captures a more complex property profile to guide generation of new molecules into the desired property space. These concepts combined with medicinal chemistry inspired structure transformations were shown to produce novel prioritized proposals following such a multidimensional optimization Scheme.
3.3.7.Reward Functions in Latent Chemical Space
- Autoencoder based design approaches work in a latent chemical space. In this setting, reward functions can be implemented into the latent space directly to predict properties for latent continuous vector representations.
3.3.8.3D-Docking Based Reward Functions
- The major drawback of docking is the time required for scoring a new proposal to give feedback to the design agent.
- In particular, docking combined with machine learning has the potential to speed up this process for use in AI-based design.
3.4.Chemical Feasibility
- One class of approaches compares molecular fragments of the molecule in question with fragments from synthesized molecules. A popular approach in this category is the synthetic accessibility score called SAScore.
- The synthetic Bayesian accessibility, SYBA, trains a Bayes classifier from easy to synthesize compounds, collected from the ZINC database, and hard to synthesize molecules, which are artificially generated.
- The SCScore builds on the complexity of the synthetic transformation. A deep feed-forward neural network was trained on 22 million pairs of reactants and products based on the idea that, on average, a chemical reaction typically increases the complexity of a molecule. Retrosynthetic analysis was taken a step further in the retrosynthetic accessibility score, the RAScore.
3.5.Integration of Scoring Functions in Internal Workflows
- Reinforcement learning provides a robust framework for AI-based de novo design, in which a molecule generator is guided by different reward functions. As reward functions, we have integrated the following scoring functions to guide reinforcement learning:
- Physicochemical property scoring using corresponding descrip- tors in RDKit.
- 2D similarity-based scoring using Tanimoto coefficients to the starting molecule based on the ECFP6 or other fingerprints from RDKit.
- 3D shape similarity scoring to the starting molecule in 3D using ROCS-3D shape similarity.
- 2D-QSAR-models trained on target activity data using Cubist regression trees or graph convolutional neural networks using the DeepChem library.
- A combined score based on addition of individual scoring terms listed above for properties, 2D fingerprints, 3D similarity, and 2D-QSAR.
- A combined score based on multiplication of individual scoring terms.
- Additionally, we tailor the search of the chemical space by adding 2D or 3D similarity calculations into the reward function.
- Transfer learning provides an additional opportunity to bias the exploration of analogues around the actual chemical series.
- Sufficient training and test sets should be selected either randomly or by statistical design. We prefer the latter approach using the RDKit implementation of the MinMax algorithm as efficient approach for picking diverse subsets from larger sets.
- An important step is to use novel data during project progression for model validation, a method known as time-split validation.
3.6.Compound Selection and Filtering
- There are, however, additional aspects to consider, when analyzing AI-based de novo design results.
- First, the synthetic accessibility of design proposals is of critical importance for their realization in the chemical laboratory. Simple estimates include the number of chiral centers, complex ring systems and availability of motifs in internal building block repositories or vendor catalogues and are routinely added to focus the selected set of com- pounds into synthetically desired regions.
- Next, the quest for novelty is addressed by 2D similarity searches in a project or corporate database and in a database of patented or reported compounds in the scientific literature for a particular project.
3.7.A Practical Example: AI-Design at Work

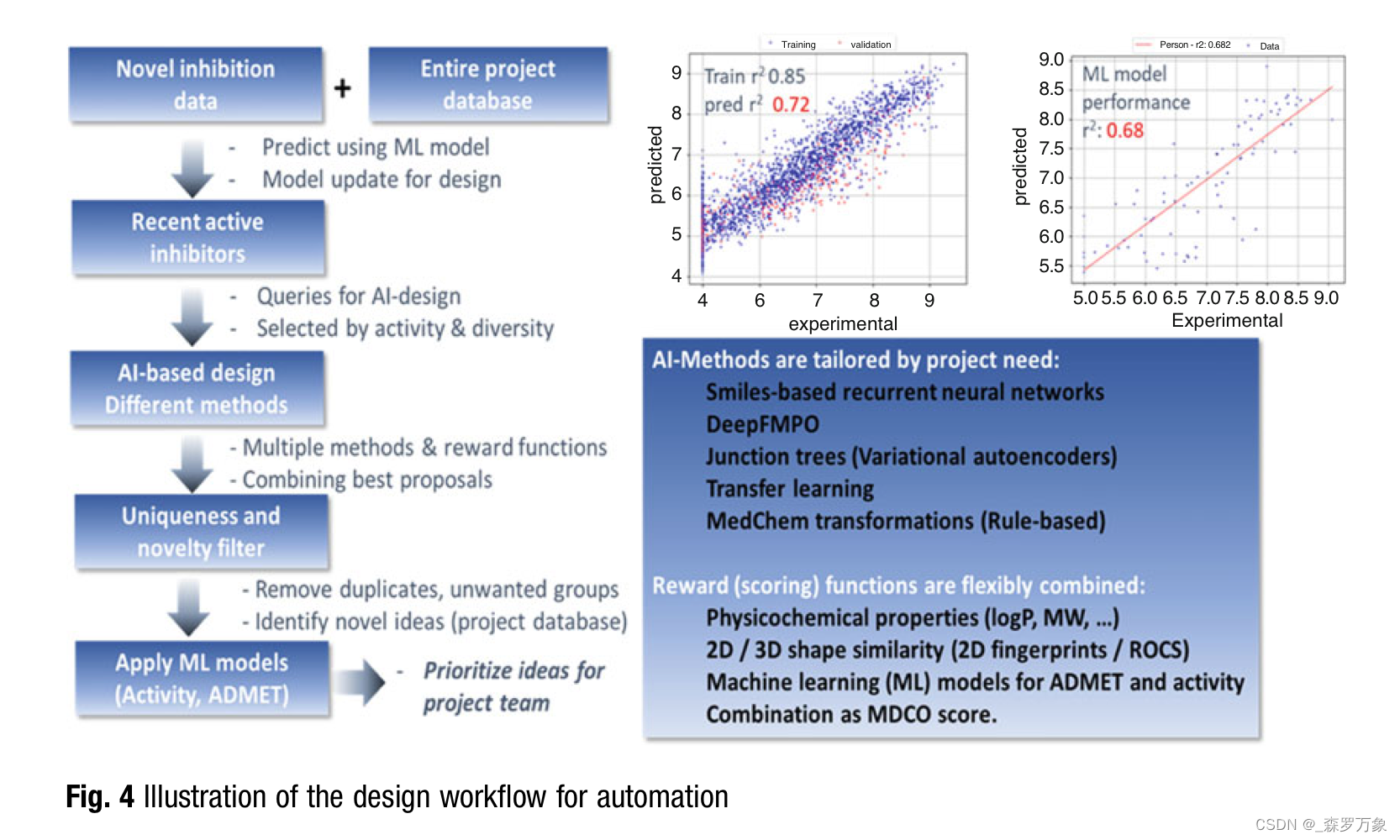

4.Summary
-
Chemists need to consider the outcome from such a statistical model to capture essential SAR knowledge for the next round of design in a project.
-
To this end, a group at GSK evaluated chemical structures from three AI-design algorithms. Significant differences were observed between these molecule generators in three different performance tests, highlighting the necessity to select the appro- priate design engine for the problem at hand. These tests include:
- the ability to reproduce ideas from an experienced medicinal chemistry team;
- the ranking of novel AI-generated ideas by medicinal chemists;
- the generation of relevant structures in legacy discovery projects starting from a single seed molecule.
















 778
778











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











