







目录
综述
在癌症预后预测的研究领域,该文章创新性地运用去噪自编码器整合多组学数据,并借助其隐层所提取的特征来评估患者的患病风险。研究人员针对来自 TCGA 数据库的15种癌症展开了全面实验,结果显示,相较于过往的方法,此方法在关键评估指标 C-index(CI值)上表现卓越,平均提升幅度达到了6.5% 。
考虑到实际应用场景中,获取多组学数据存在诸多困难,研究团队进一步探索优化方案。他们尝试仅利用mRNA数据,通过训练 XGBoost 模型来拟合风险估计值。令人惊喜的是,经测试该模型的平均 C-index 值达到了0.627。这一成果不仅为癌症预后预测提供了更具可行性的方法,也为后续相关研究开辟了新的思路。
研究背景
癌症是一种复杂的疾病,涉及基因与环境的一系列相互作用。临床研究发现,即便同为一种癌症类型的患者,其癌症预后也存在显著差异,这严重阻碍了有效癌症疗法的发展。因此,依据基因组信息精准区分高风险和低风险患者至关重要。目前,基于基因组学信息评估癌症预后风险的研究众多,其中基因表达(mRNA)数据最为常用。随着下一代测序技术的发展,DNA 甲基化、miRNA、拷贝数变异(CNV)等多种其他类型的基因组数据也可获取,整合多组学数据有助于更全面地捕捉癌症预后预测中的复杂性。
癌症基因组图谱(TCGA)组织对多种癌症的多组学数据进行了测序,推动了相关统计方法的发展,如基于稀疏偏最小二乘判别分析、无监督多核框架、语法进化神经网络等方法被用于多组学数据的整合与分析。然而,这些传统线性方法在处理多组学数据的高维异质性变量时,难以有效捕捉代表性特征。
深度学习技术在处理非线性问题方面表现出色,一些基于深度学习的方法也被应用于癌症生存分析,如 DL-Cox、使用自动编码器(Autoencoder)提取特征的方法等。但 Autoencoder 在学习代表性特征时易受数据噪声影响,且以往研究多聚焦于单一癌症类型,缺乏对泛癌的综合测试。
在这样的背景下,该论文提出了一种新的框架,以更有效地整合多组学数据,实现精准的癌症预后预测。
研究方法
数据预处理
数据获取
从TCGA数据库下载癌症数据集需要大量的工作去处理样本和四种多组学数据。 UCSC Xena 网站提供处理好的样本和组学数据,所以通过 UCSC Xena 获取样本数据与四种组学数据。这里以 LUSC 癌症为例。
在 UCSC Xena 数据库中找到 LUSC 癌症数据库后,点击进去里面包含了各种类型的数据,包括了我们要的mRNA、miRNA、DNA甲基化以及CNV数据。点击对应链接并下载即可获取对应数据。
数据预处理
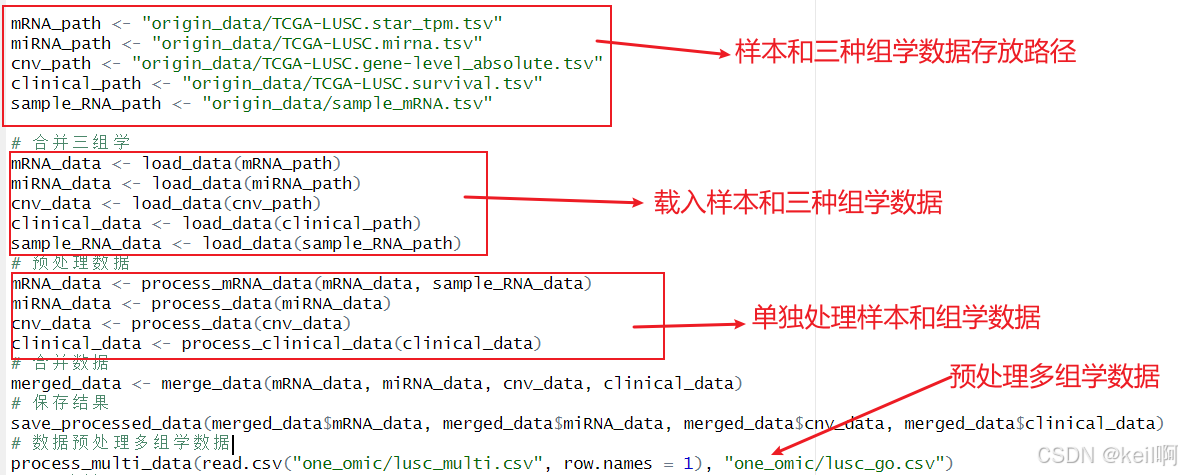
在进行缺失值处理之前,我们需要对上述四种数据进行整合。首先,执行load_data载入样本信息与三种组学数据(由于本人电脑内存不够所以仅尝试了三种组学)。接着,执行process_data对每种组学数据进行单独处理,执行 process_mRNA_data对mRNA单独进行处理,函数内执行基因ID与基因Symbol的转换以及筛选基因类型为mRNA的基因,执行rocess_clinical_data对临床数据进行筛选仅保留样本的生存时间和生存状态。最后,执行merge_data对上述样本数据和组学数据进行整合,最终会生成处理好的单组学文件、临床信息文件和多组学文件。
缺失值处理的过程包括,先排除在超过 20% 患者中缺失的特征,接着排除缺失超过 20% 剩余多组学特征的患者样本,再剔除未删失样本少于 50 个的癌症数据集。对于剩余样本的缺失值,使用 R 包imputeMissings基于中位数进行填补。在这里,执行process_multi_data将会完成上述缺失值处理的全过程并生成LUSC_go.csv文件。
![]()
框架结构拆解
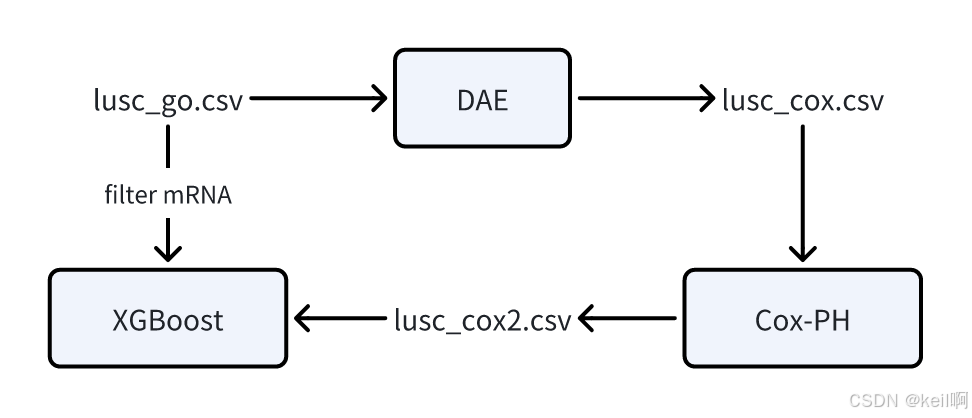
论文框架如下图所示,共有三个模块,分别为自编码器模块、Cox-PH单因素方差分析模块和XGBoost模块。
去噪自编码器模块
与普通自编码器相比,DAE通过在输入数据中添加噪声(如随机噪声或部分数据丢失),并训练模型恢复原始数据,从而增强模型对数据噪声的鲁棒性。这使得模型在面对实际数据中的噪声和不完整信息时,能够更稳定地工作。
DAE是整个预测框架的第一步,负责将多组学数据(如mRNA、miRNA、DNA甲基化和CNV)整合并压缩为低维特征。这些低维特征随后被输入到Cox模型中用于风险预测。
Cox-PH模块
Cox模型是一种经典的生存分析模型,用于评估患者的生存风险。它通过分析患者的特征(如基因表达水平)与生存时间之间的关系,计算每个患者的相对风险。在论文中,Cox模型使用DAE提取的低维特征作为输入,预测患者的生存风险。
Cox模型的一个优点是具有一定的可解释性。通过分析模型的系数(β),可以了解哪些特征对生存风险的影响更大,从而为生物学解释和临床应用提供依据。
Cox模型是整个预测框架的第二步,它接收DAE提取的低维特征,并输出患者的生存风险评分。这些风险评分用于将患者分为高风险和低风险亚组。
在这里执行process_cox_file,传入临床信息与cox分析结果文件,以及保存文件即可进行cox分析,接着执行perform_anova_and_plot绘图。

XGBoost模块
XGBoost是一种基于梯度提升的机器学习算法,能够高效地处理大规模数据,并且具有很强的特征选择能力。在论文中,XGBoost用于从mRNA数据中选择关键基因特征,构建轻量级的预测模型(DCAP-XGB)。这种轻量级模型不仅能够提高模型的可解释性,还能够在实际应用中更高效地运行。
XGBoost通过评估每个特征对模型性能的贡献,自动选择最重要的特征。这使得模型能够专注于与癌症预后最相关的基因,从而提高预测性能。
XGBoost是框架的第三步,它接收DAE和Cox模型的输出(即患者的生存风险评分),并基于mRNA数据选择关键基因特征,构建轻量级的预测模型。这些轻量级模型可以用于独立数据集的验证,并用于发现与癌症预后相关的生物标志物。
通过执行perform_analysis拟合mRNA与生存分析评分构建轻量级的预测模型

实验结果
多组学数据预测效果
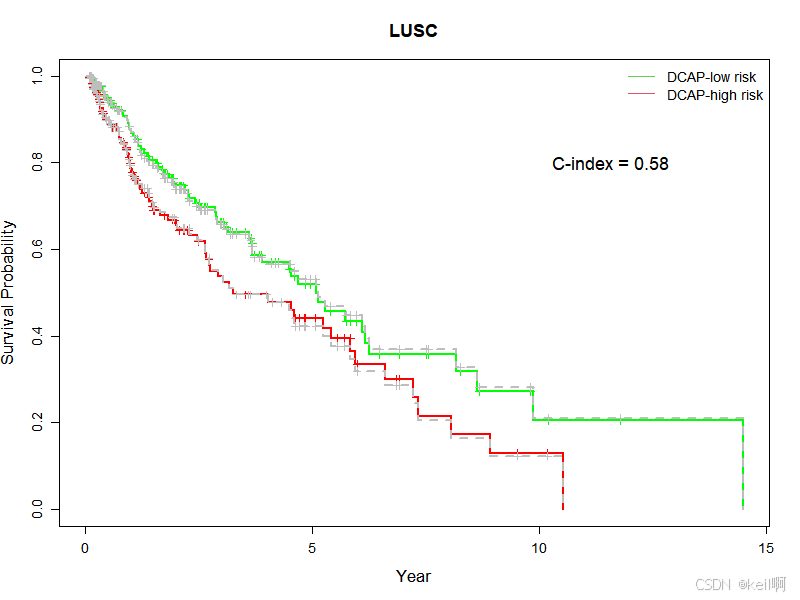
在LUSC癌症数据集上,通过mRNA、miRNA、CNV三种组学数据进行生存分析,CI值为0.58。而原论文中,使用四种组学数据进行生存分析的CI值为0.597,原论文中指出DNA甲基化数据对于CI值的影响在四种组学数据中排第三,所以可能是由于缺失了一种组学数据导致CI值下降了1.7个百分比。
DCAP-XGB预测效果
XGBoost的CI值相较四种组学数据得出的CI值少了2个百分比。但是XGBoost的输入仅有mRNA数据。
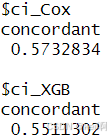
源代码
本人对源代码进行整理并重新封装,DAE采用pytorch重新实现,数据预处理全过程封装为函数并保存为main.R文件,源代码下载链接
总结归纳
该篇论文使用去噪自编码器(DAE)处理高维多组学数据,获取隐层特征,减少数据噪声影响,增强模型鲁棒性。将 DAE 提取的特征输入 Cox 比例风险模型,评估患者的癌症风险。针对多组学数据获取困难的问题,使用 mRNA 数据训练 XGboost 模型拟合风险,提高模型的可解释性和临床适用性。
通过实验分析各类型组学数据对预后预测的贡献,发现 mRNA 数据的贡献最大,单独使用时平均 C-index 值为 0.628,而 CNV 数据贡献最小。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









