







目录
综述
该论文提出了一种基于堆叠自编码器的多组学数据整合方法 SAEsurv-net,用于癌症生存预测。通过两阶段降维和堆叠自编码器模型,有效解决了多组学数据的高维和异质性问题,实验结果表明该方法在多个癌症数据集上表现优于现有方法。
研究背景
多组学数据整合的重要性
癌症的发生和发展涉及基因组、转录组、表观基因组等多个层面的变化,整合多组学数据能更全面地揭示癌症机制,提高生存预测准确性。
挑战
维度灾难
多组学数据特征维度极高(如基因表达数据有 6 万 + 特征),易导致过拟合和计算复杂度问题。
数据异质性
不同组学数据在特征数量、分布、尺度等方面存在差异,整合难度大。
现有方法的局限性
早期整合
将数据直接进行拼接,可能会导致维度爆炸和噪声增加。
晚期整合
将模型的结果进行融合,这将无法捕捉组学间交互作用。
中间整合
对模型进行联合训练,难以处理高维输入。
研究方法
数据获取
-
从TCGA 获取癌症的基因表达、CNV 和临床数据。
-
从UCSC Xena网站下载整理过的癌症的基因表达、CNV 和临床数据。
数据预处理
归一化
基因表达数据进行 log2 转换和标准化,临床数据处理为独热编码或标准化
临床数据
对临床数据中的非数值列进行独热编码,数值列进行归一化处理。
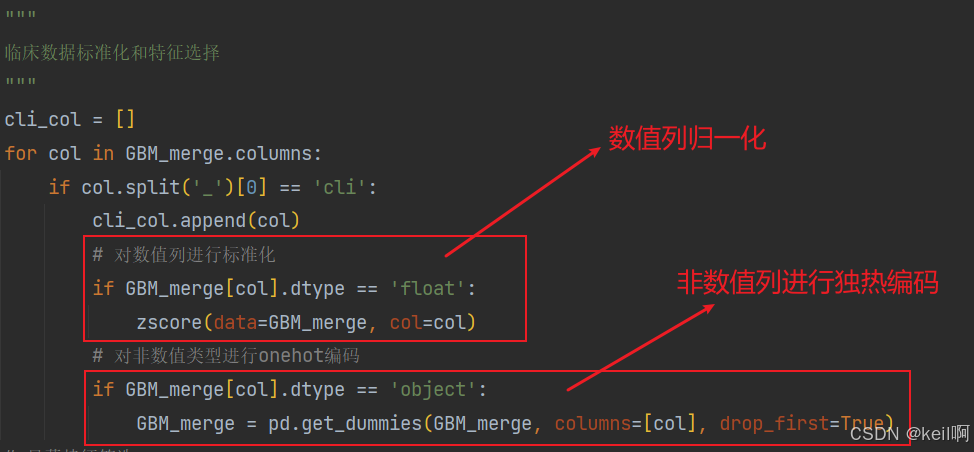
基因表达
对基因表达的特征列进行归一化处理。
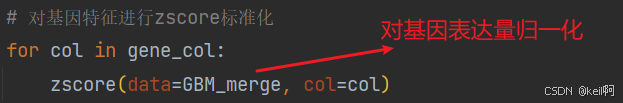
特征筛选
临床数据
筛选p值小于0.05的特征

CNV
对于CNV的筛选需要有三步,第一步是根据方差进行筛选,筛选方差不为零的特征列;第二步是筛选重置值的特征列;第三步是筛选高表达的特征列。
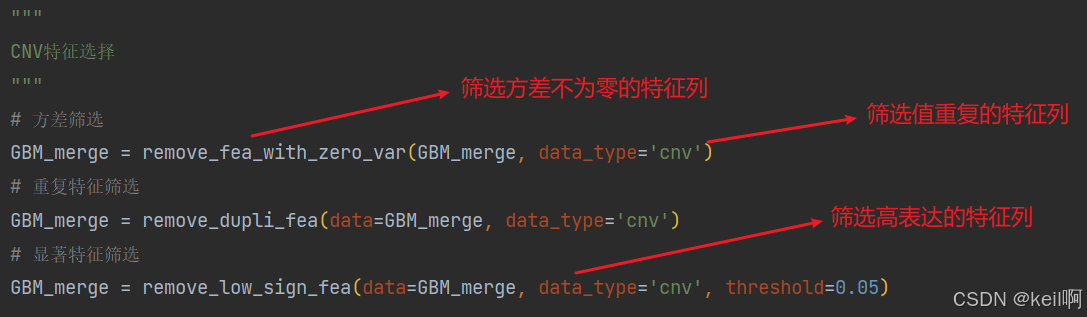
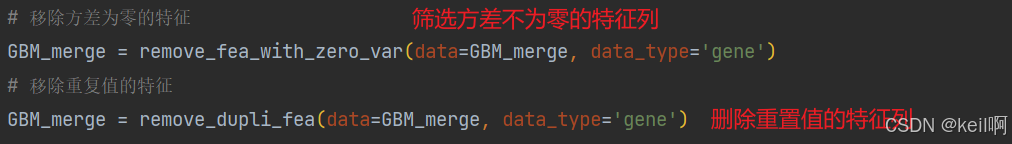
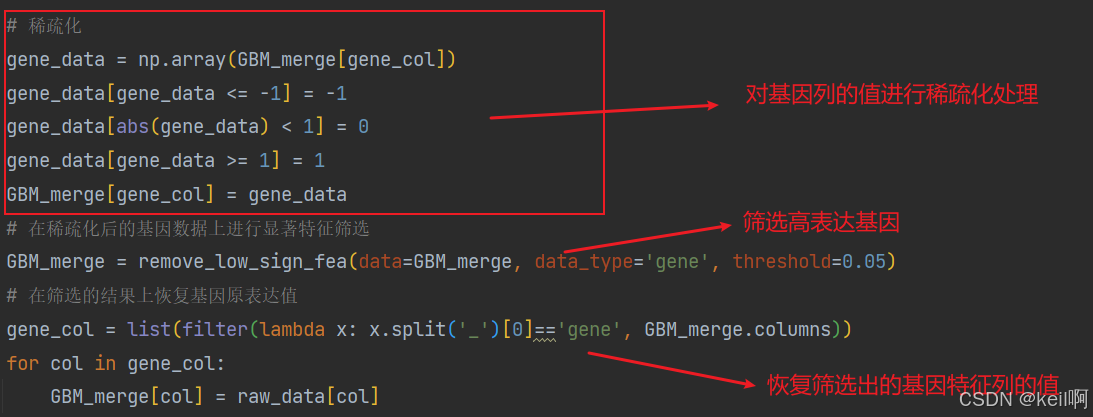
基因表达
对基因表达的特征也需要进行三步筛选,相较于CNV,在筛选高表达特征列前对特征值进行稀疏化处理,筛选后恢复特征值。
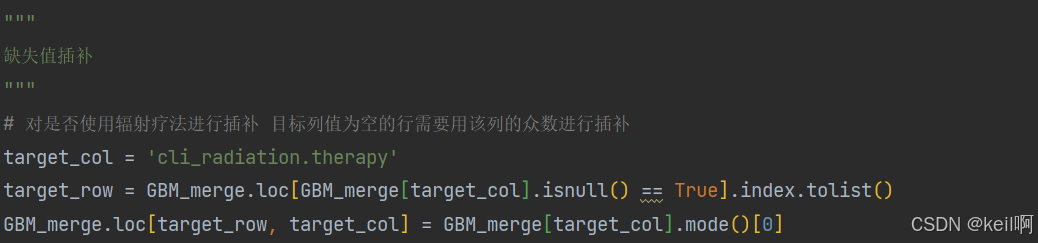
缺失值插补
这里有一个列名为是否使用辐射疗法的临床数据特征列,其列值中存在缺失值,这里采用众数插补法,对缺失值进行插补。
两阶段降维与整合
第一阶段
对每个组学数据独立进行上述数据预处理过程。
第二阶段
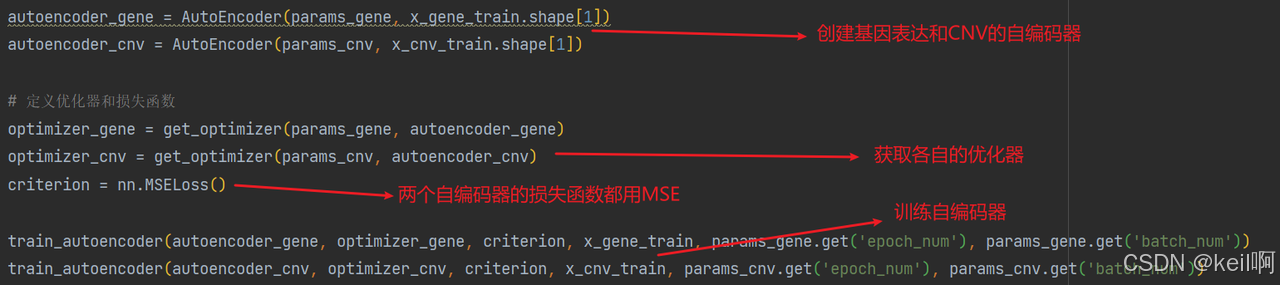
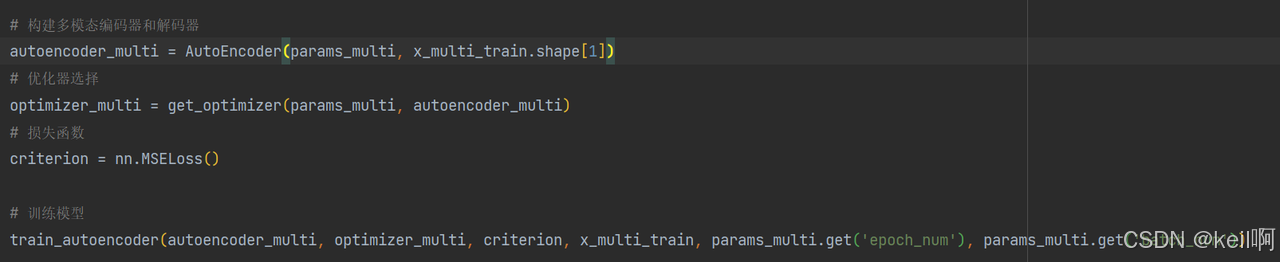
堆叠自编码器
-
对基因表达和 CNV 数据分别训练自编码器,提取低维特征。
-
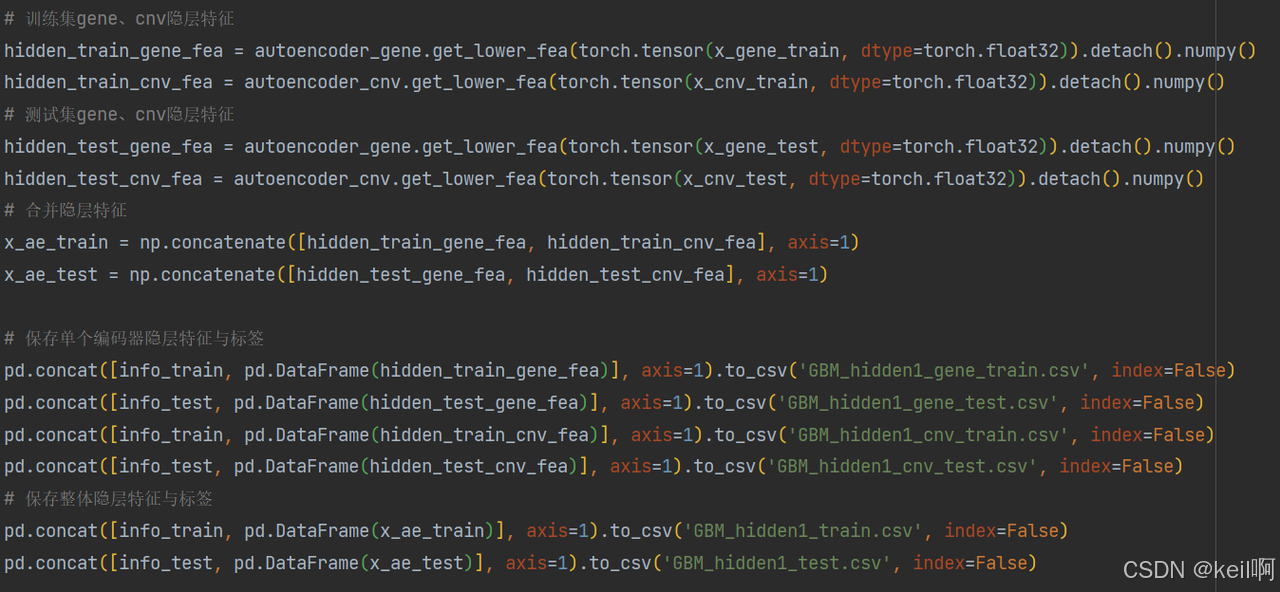
将两组学特征拼接并与临床信息拼接
-
训练第二个自编码器,生成跨组学整合特征
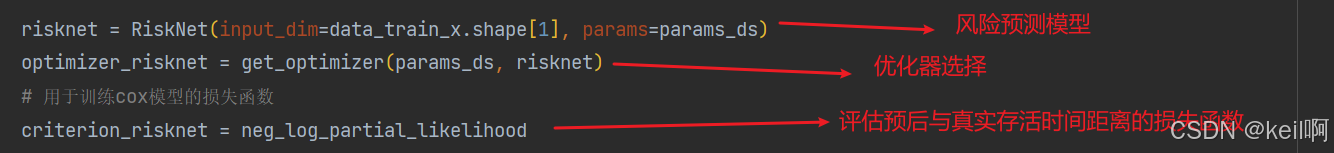
风险预测模型
将跨组学特征作为输入,训练风险模型预测生存风险(Prognostic Index, PI)。
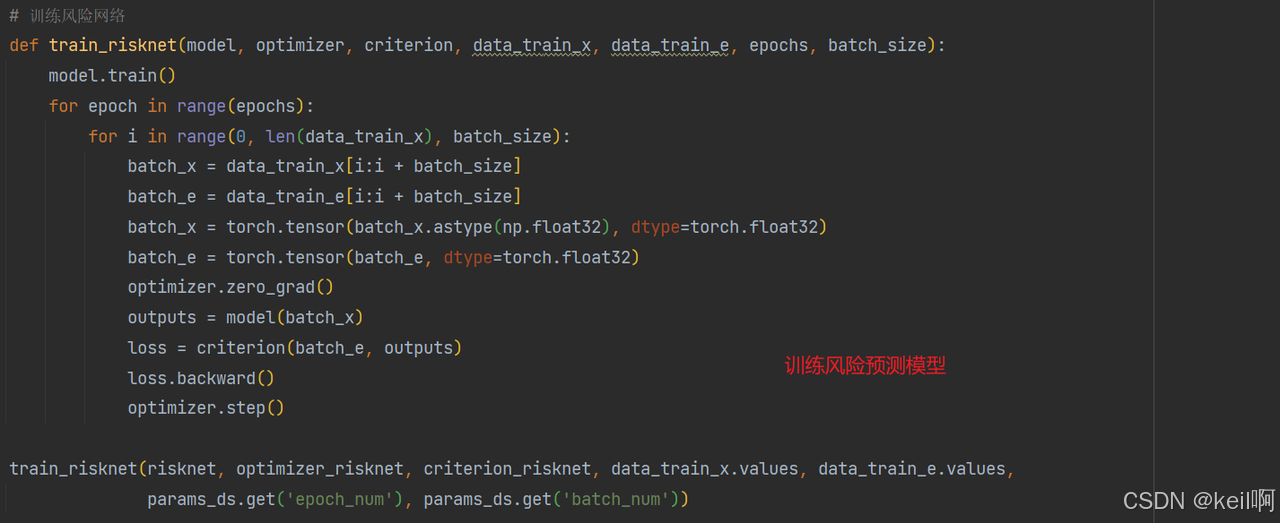
训练风险预测模型
实验结果
验证并测试风险预测模型
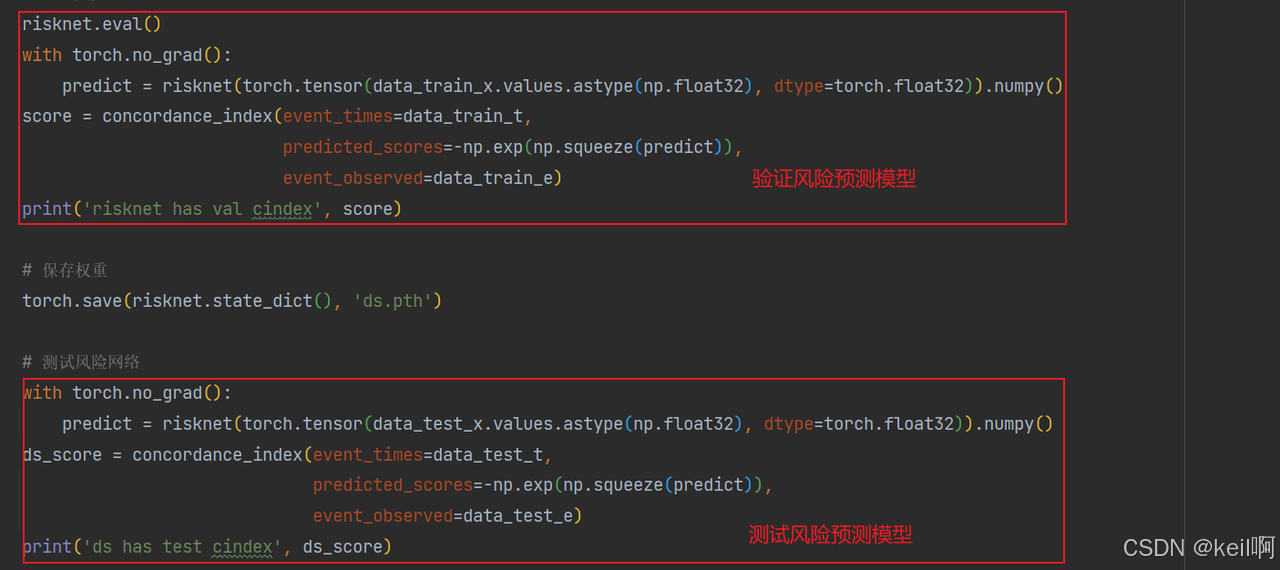
结果
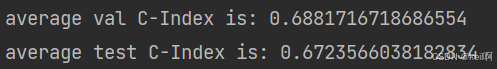
平均的验证CI值为68.81%,平均的测试CI值为67.24%,最大的验证CI值为77.25%,最大的验证CI值为65.28%,与原论文中62.12%相比还略高一些😀
源码
本人将原来的代码进行修改,并采用Pytorch重新实现,主要分为两个文件,data_preprocess.py文件用于对数据进行预处理,train.py文件用于两阶段训练和风险预测模型的评估。代码下载链接
结论
该论文提出了两阶段SAE框架以及一个基于深度学习的风险预测模型,有效的解决了高维和异质性问题,通过非线性特征学习提升了模型的鲁棒性。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









