









首先是PTM的介绍:
参考:https://en.wikipedia.org/wiki/Post-translational_modification

蛋白质的翻译后修饰(PTM)通过改变氨基酸残基的化学结构,显著影响其带电性质,从而调控蛋白质的功能、定位和相互作用。
一,如何在uniprot中查看每个蛋白质的PTM修饰信息:
还是以CTCF为例子:
https://www.uniprot.org/uniprotkb/P49711/entry#ptm_processing
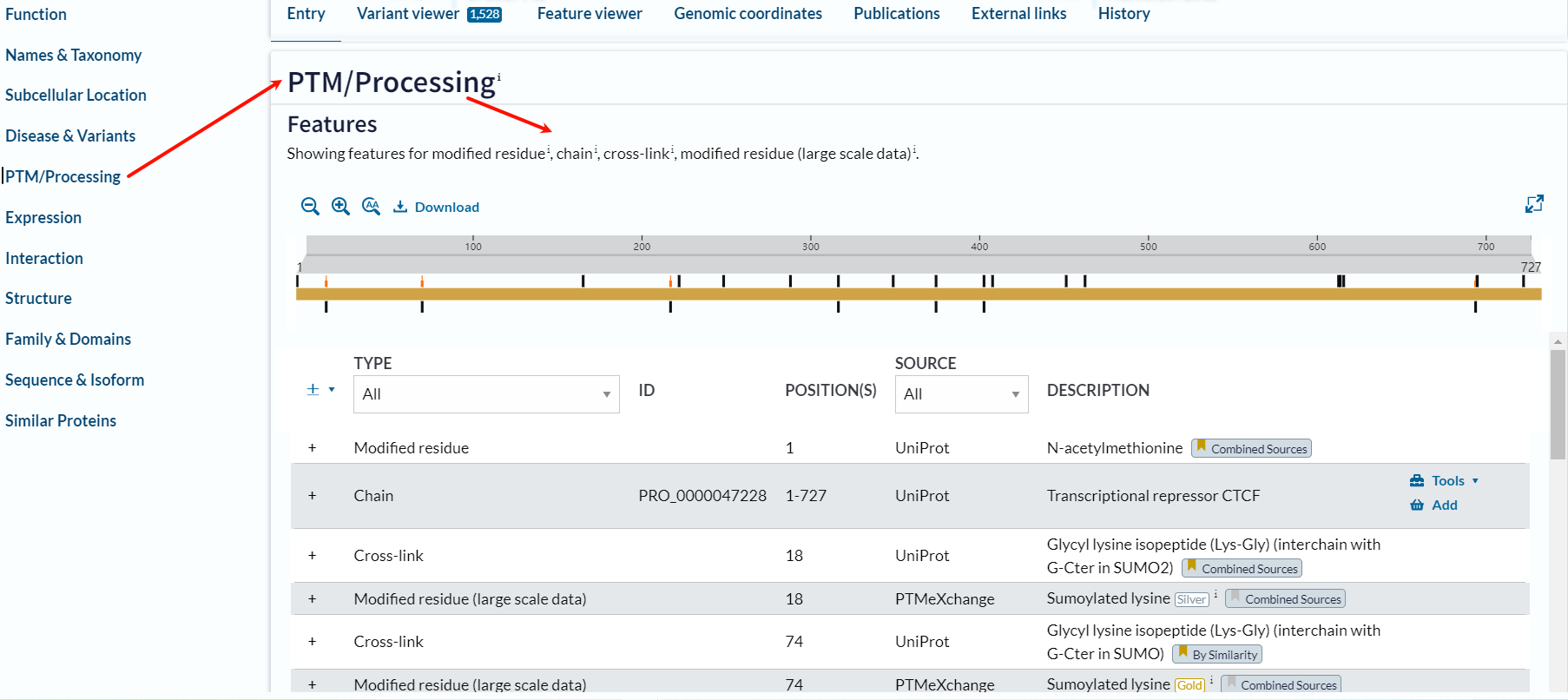
首先我们可以看到:
残基的PTM修饰信息基本上有4类:
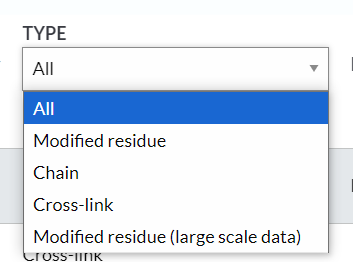
1,modified residue:最常见的
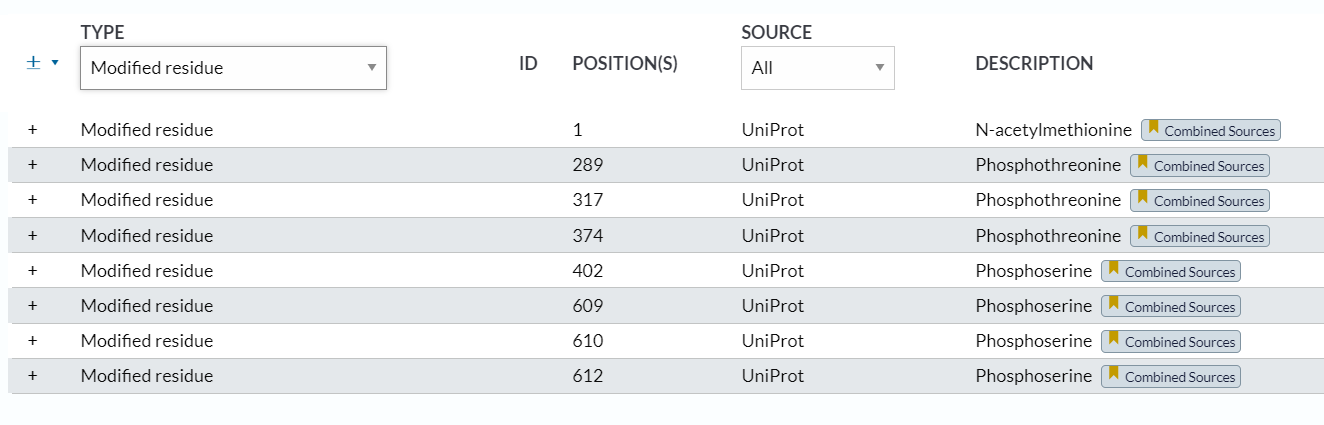
https://www.uniprot.org/help/mod_res


help界面也解释了一些常见的PTM修饰:
磷酸化:


甲基化:




乙酰化:


氨基化:


吡咯烷羧酸:


异构化:

羟基化:

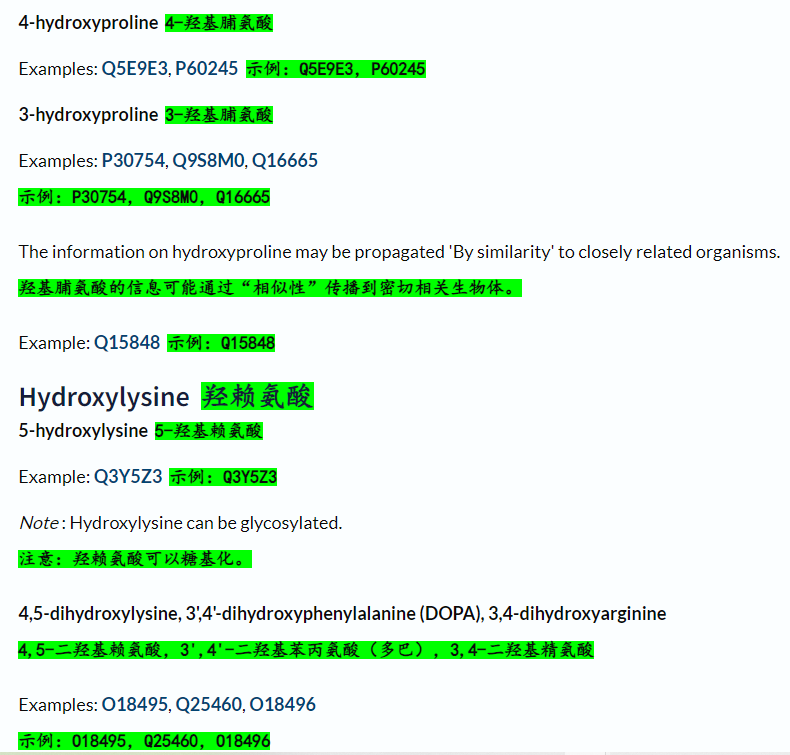

硫化:


黄素结合:

半胱氨酸氧化和硝基化:


再回过头来看:
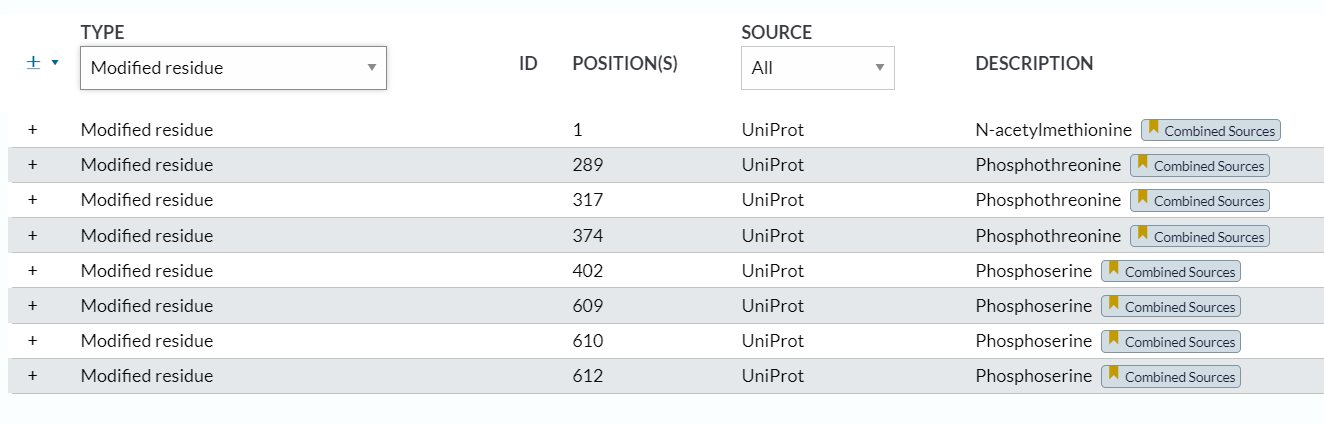
来源source都是uniprot,然后description表明是“xxx修饰xxx残基”
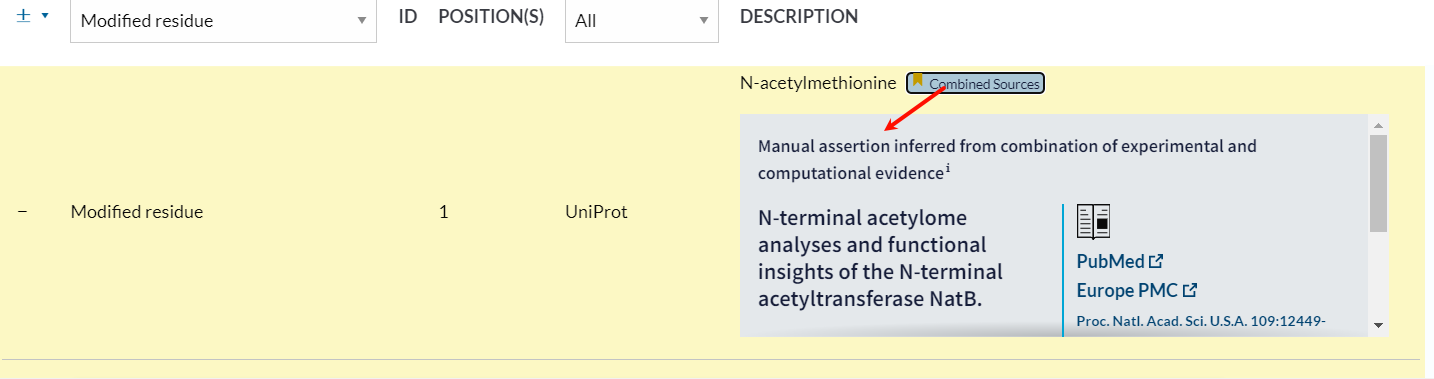
仔细点开发现有注释来源,是来自于文献,可见是人工整理的
例如:
https://www.uniprot.org/help/evidences#ECO:0007744

可以说这一部分数据就是uniprot中人工整理的,一般都是来自于文献中报道证实的修饰数据,会比较可靠
2,chain:
https://www.uniprot.org/help/evidences#ECO:0007744
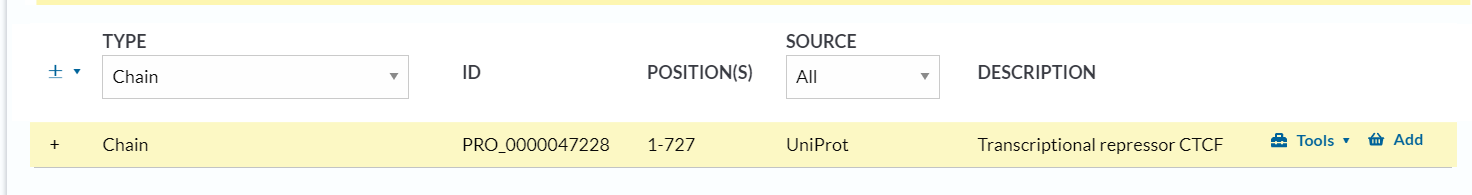
这个条目其实和我们理解的氨基酸水平上的翻译后修饰不太一样:

一般是“经过处理或蛋白酶解切割后成熟蛋白质中多肽链”那个层面上发生的广义修饰事件,不再是单个氨基酸水平上的修饰
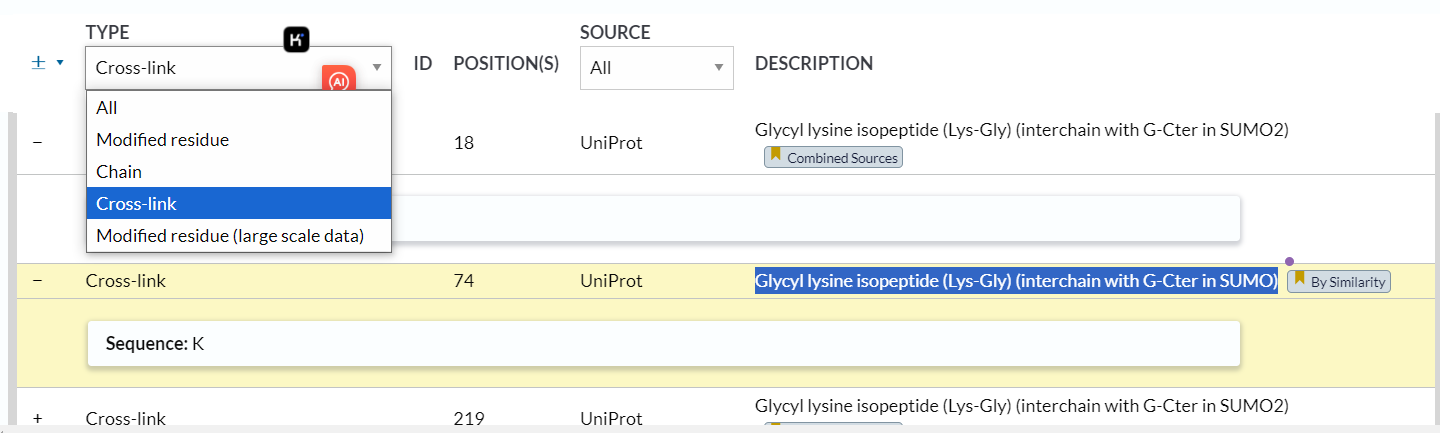
3,crosslink:
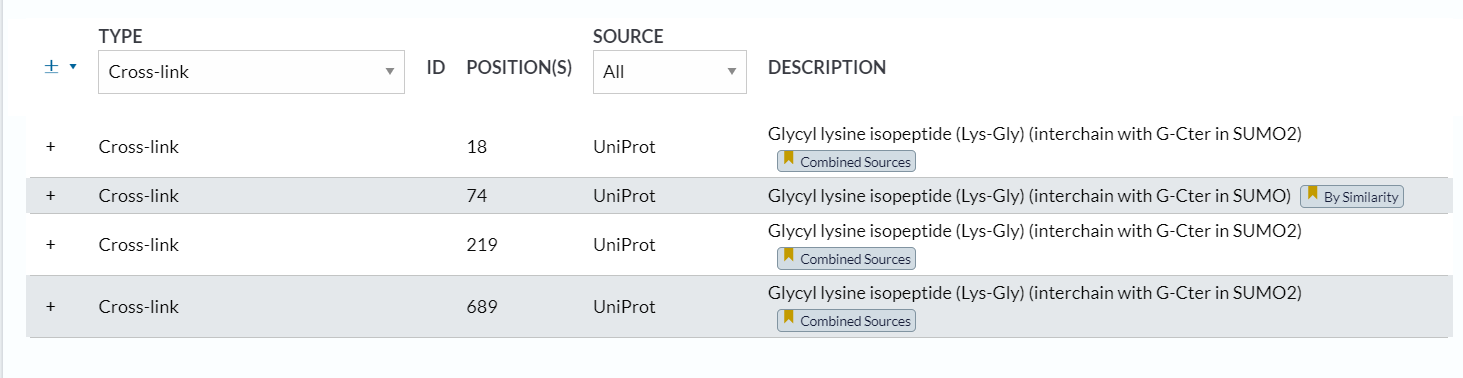
https://www.uniprot.org/help/crosslnk
其实我们可以发现描述的是chain层面上的link事件的修饰,也不是残基水平上的




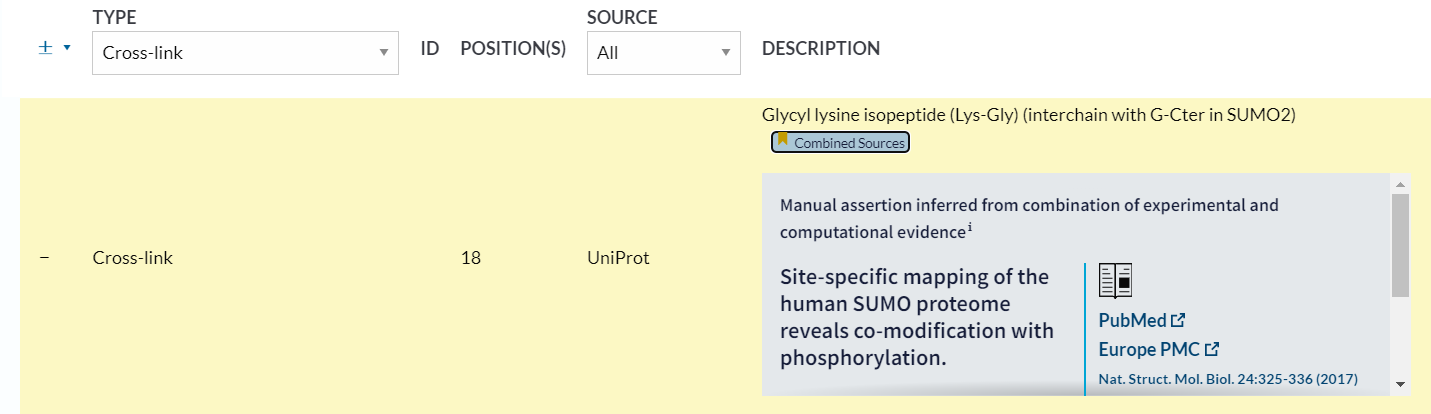
同样的,互作的修饰关系会引用文献,还是有一定可靠性的
4,modified residue(large scale data)
https://www.uniprot.org/help/mod_res_large_scale
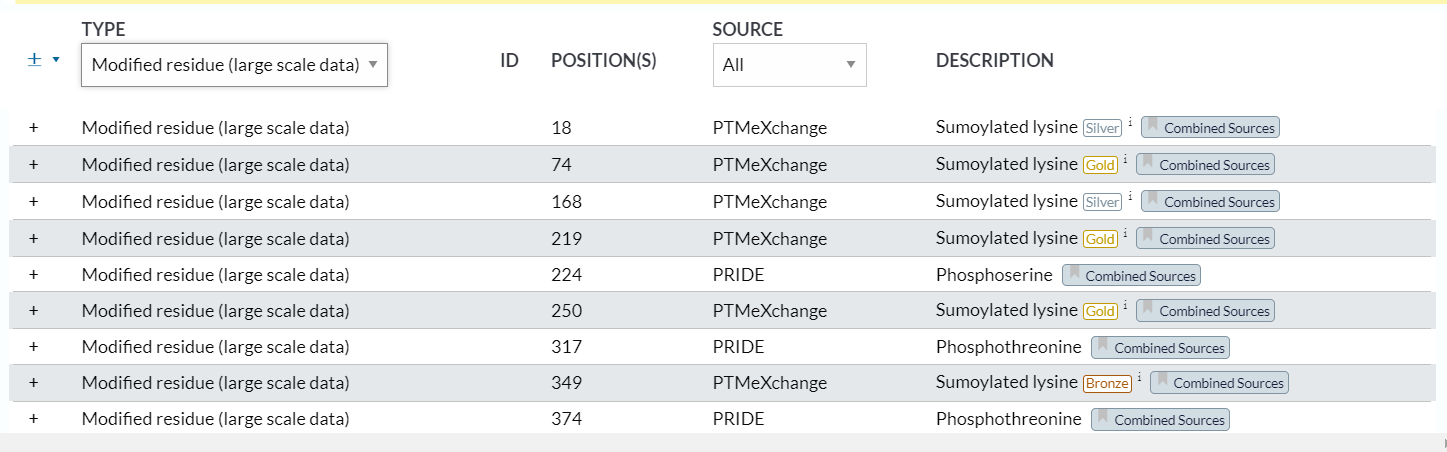

可以看到,我们能够下载的数据部分是(1)条目中的手动整理,来源是uniprot的数据,
但是还是有一些数据是额外提供的,在可下载版本里是看不到的数据,也就是常规方法无法下载的数据;
这部分数据是来自于大规模蛋白质组学项目,也就是大规模质谱项目中获取的数据信息:
数据主要来源于两个项目:

我们可以简单理解为除了人工注释的基因组数据之外,还有ENCODE、TCGA之类的大规模项目中的基因组数据,就这么来类比。
总之还有一大批规模的数据,没有经过人工整理,因为比较复杂;



然后其实我们也看到了:
这些大规模数据的PTM右上角有金银铜的标注
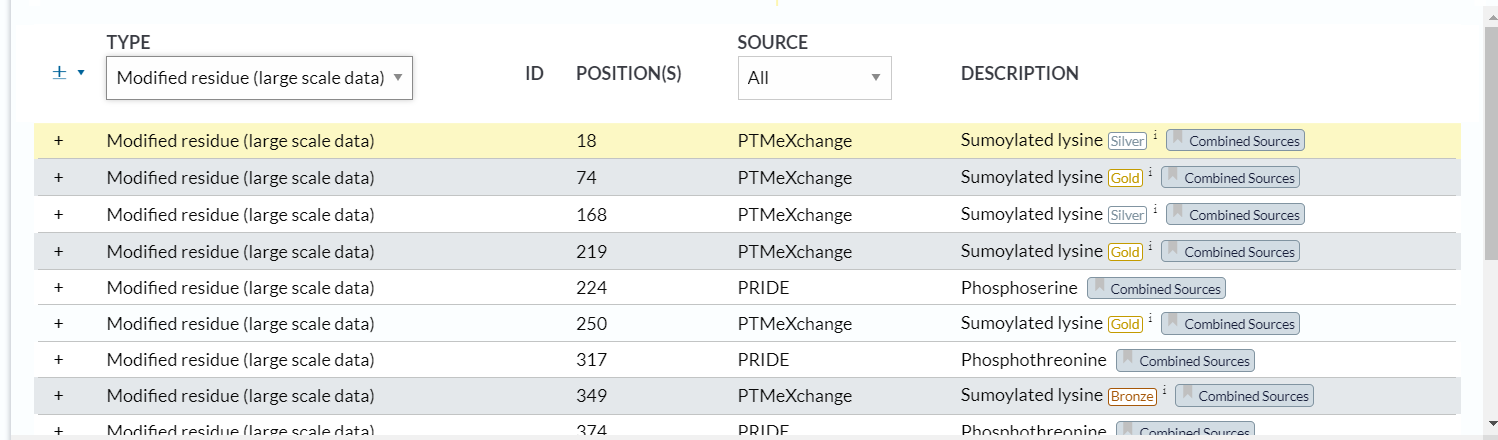


同样的,我们查看已经描述好的数据:
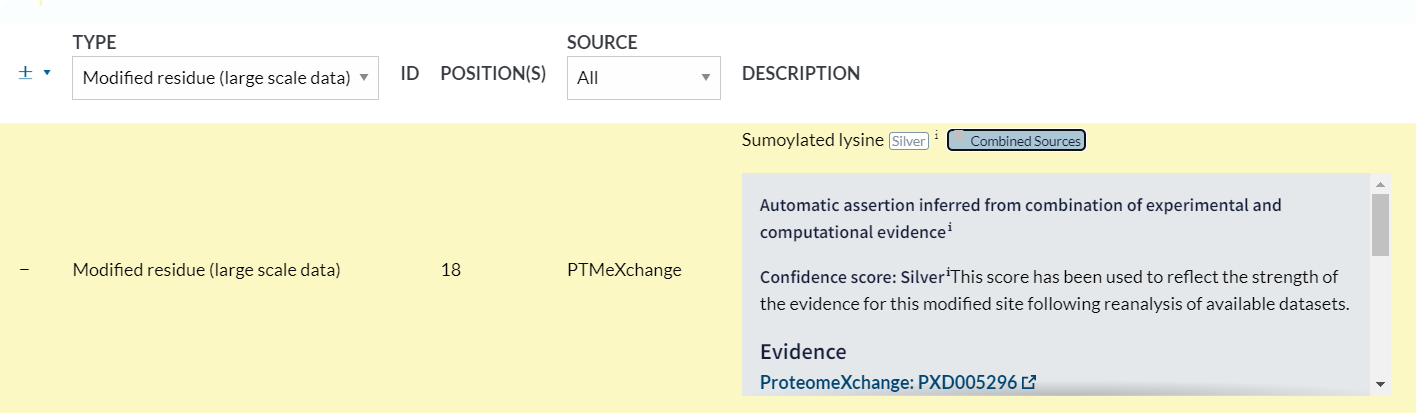
除了提及的2个大型项目,同样还是有文献引用,所以是combined

当然其实我们点进去:
https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD005296
发现就是这篇文献,所以应该是大型项目本身的文献
再看一个同理:
https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD001281
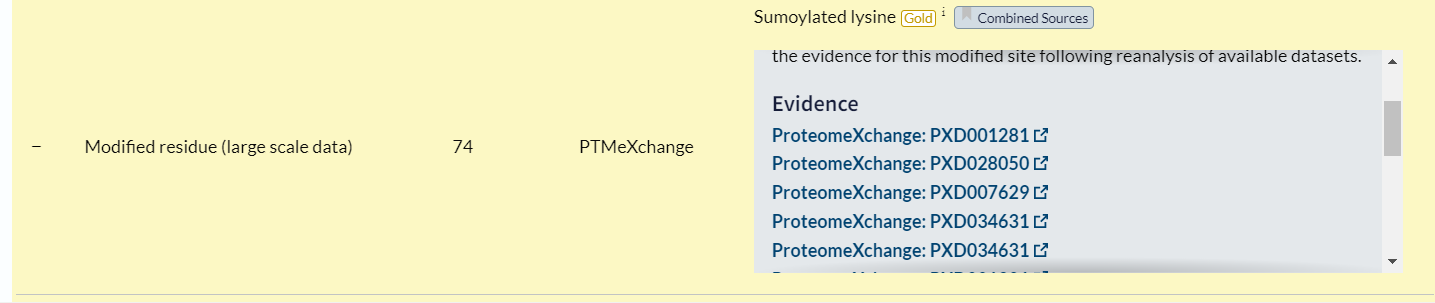


就是项目本身的文献
当然,还有一些是没有引用文献的:

——》我已经测试过了,我们平常在选定范围后的蛋白质result界面中,能够下载的PTM信息实际上只有(1)条目中的数据,是下载不了这部分大规模组学数据的修饰信息的
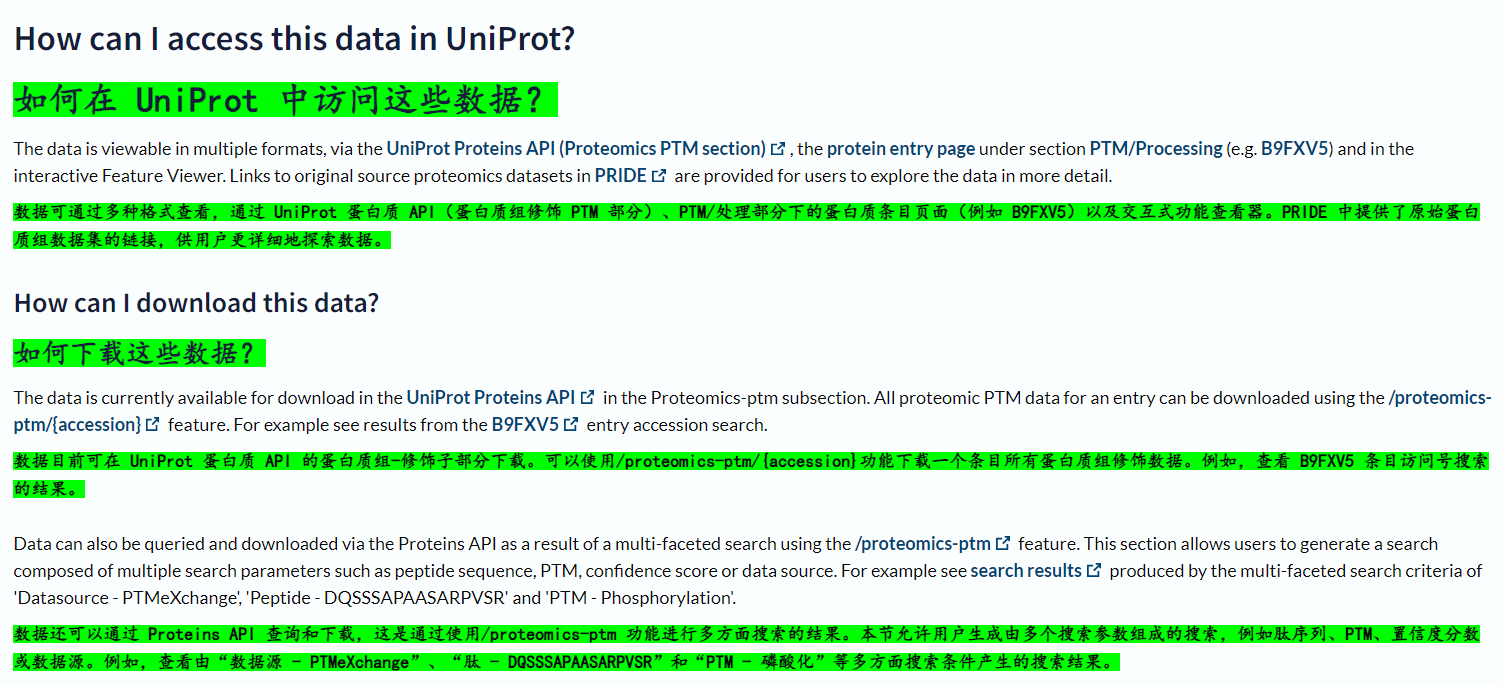
要下载的话,只能使用uniprot提供的API接口,然后自己编写python自动化脚本去获取这部分数据
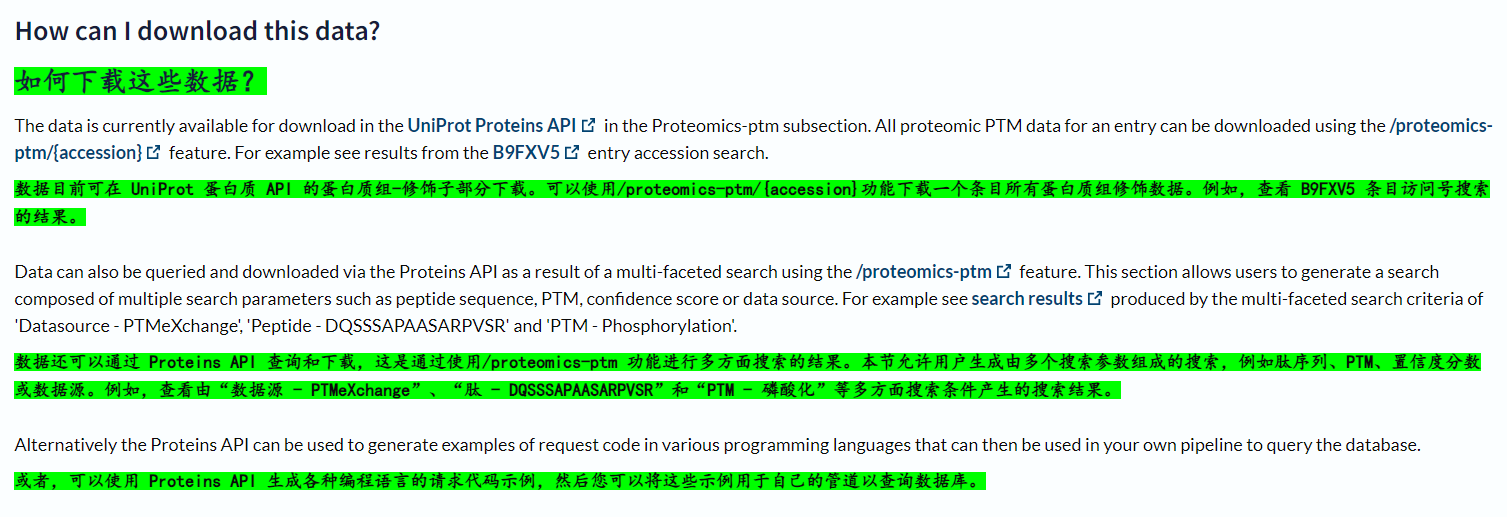
当然了,还有一种方法,就是用爬虫,直接一力降十会;
最笨的方法,就是一个一个蛋白质去搜索,然后记录在csv或者excel表格文件中
二,如何下载这些PTM修饰的数据:
1,常规的方法就是使用结果界面中的download
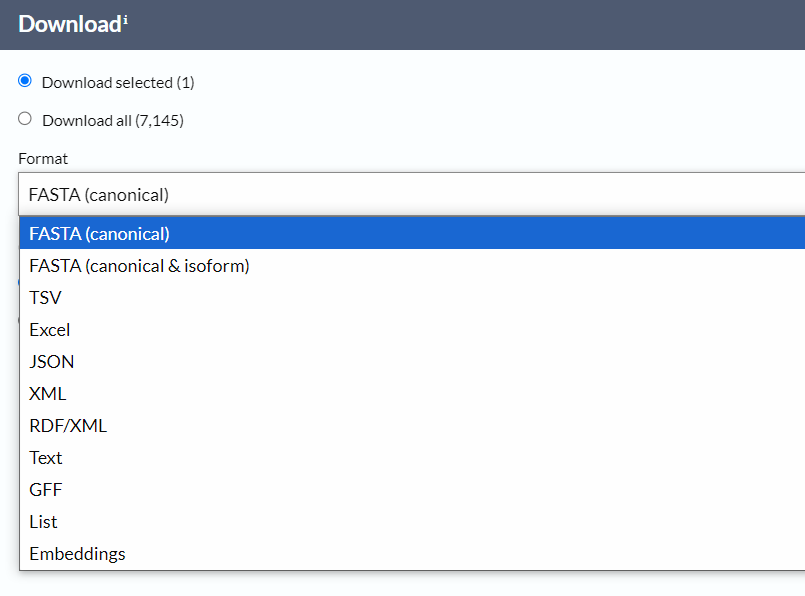
因为我们后期需要注意的是所有的修饰类型不是一起修饰的,肯定是分时期以及特征生理条件的,所以我们得经过文献校验,确定我们代码中是不是将统一并且确定的修饰位点都考虑进去了,而不是一股脑全都放上去
我此处使用的是text格式
(1)PHOSPHORYLATION 磷酸化(全大写)
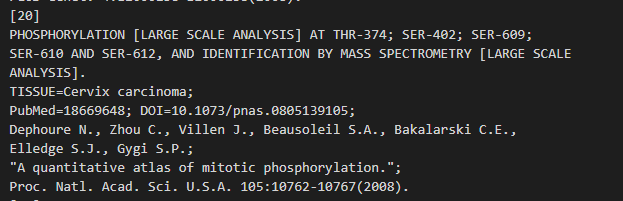
(2)ACETYLATION 乙酰化
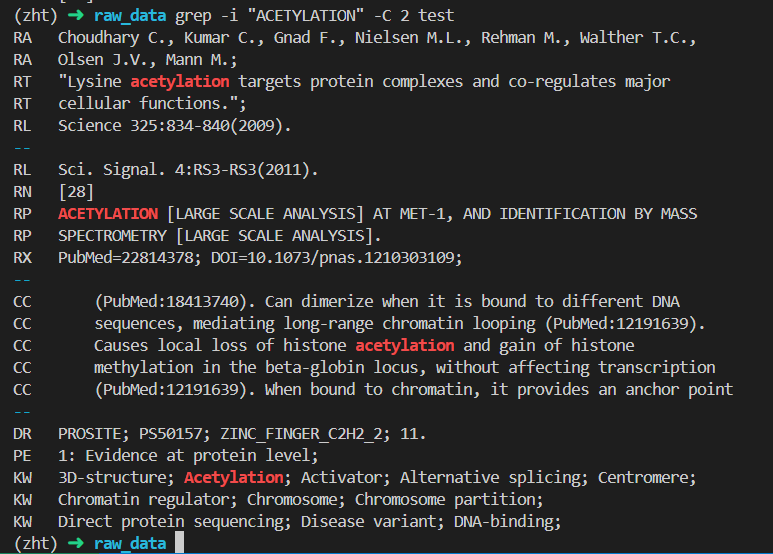
(3)SUMOYLATION SUMO化
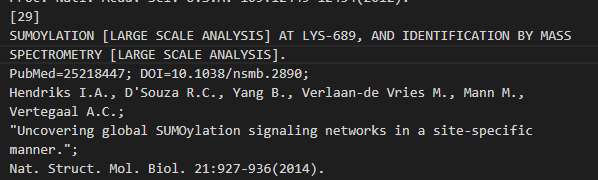
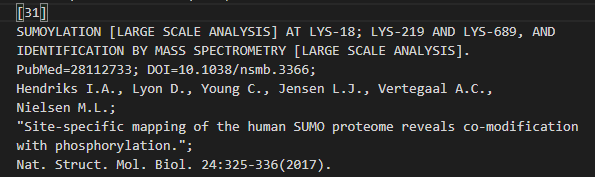
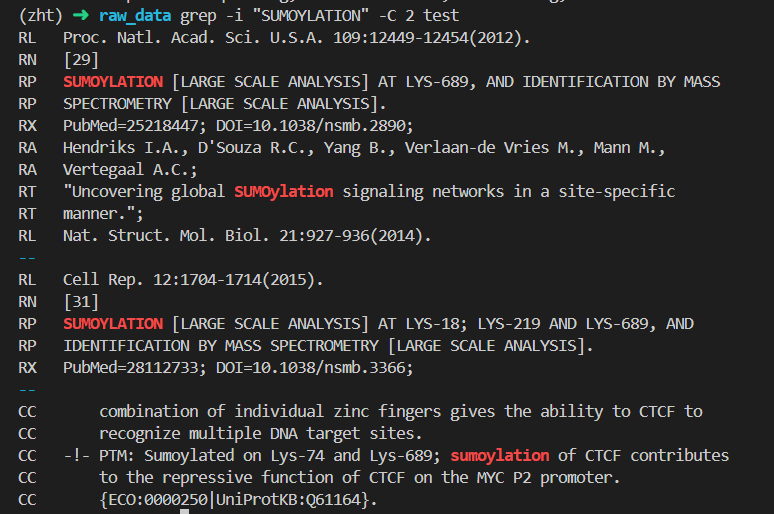
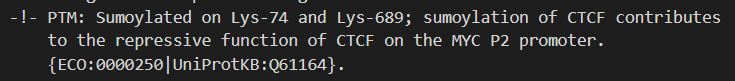
对照uniprot中的数据,
我们应该主要关注两个modified即可
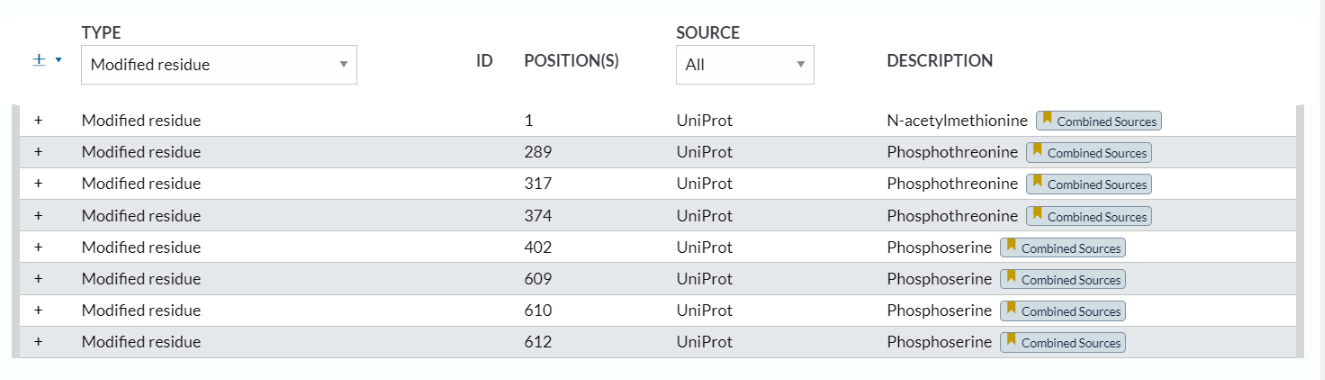
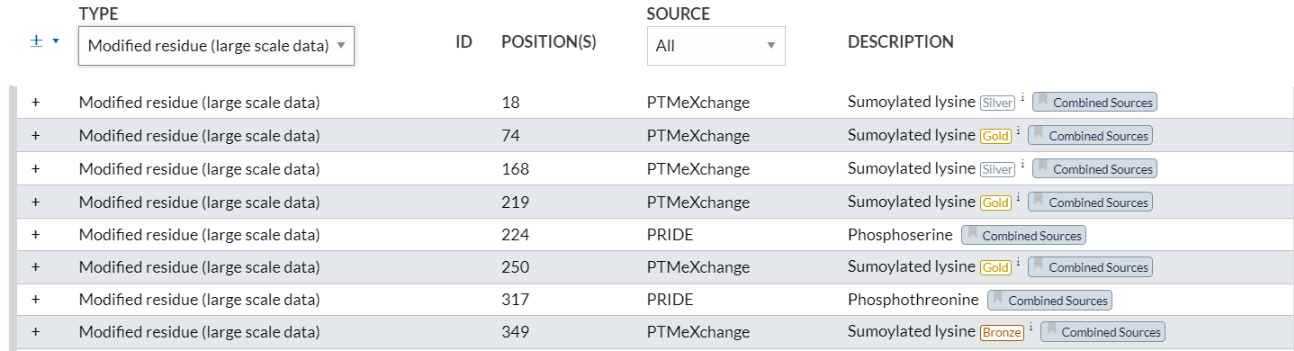
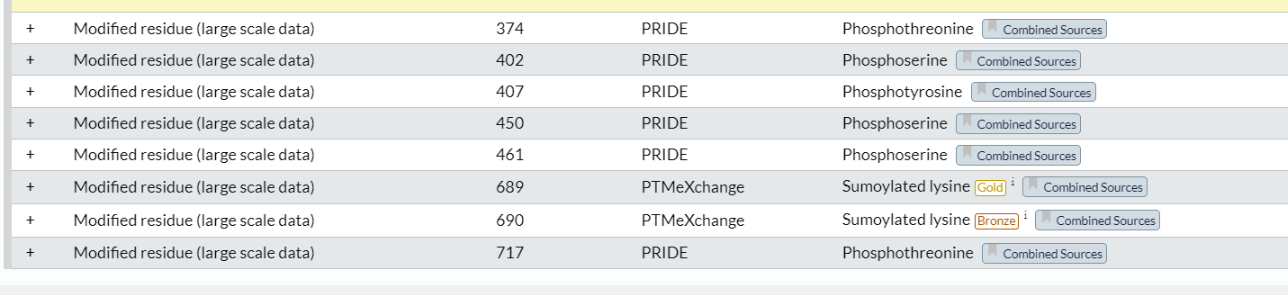
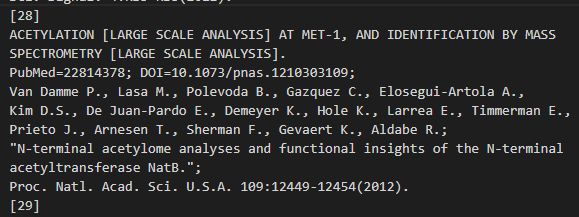
位点 1:N-acetylmethionine x-28
位点 18:Sumoylated lysine (Silver) x-31
位点 74:Sumoylated lysine (Gold)
位点 168:Sumoylated lysine (Silver)
位点 219:Sumoylated lysine (Gold) x-31
位点 224:Phosphoserine
位点 250:Sumoylated lysine (Gold)
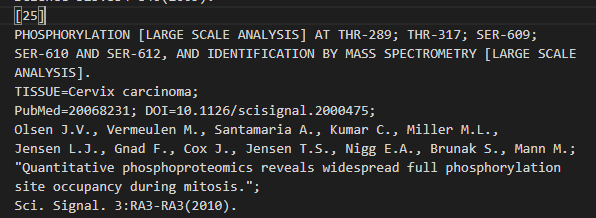
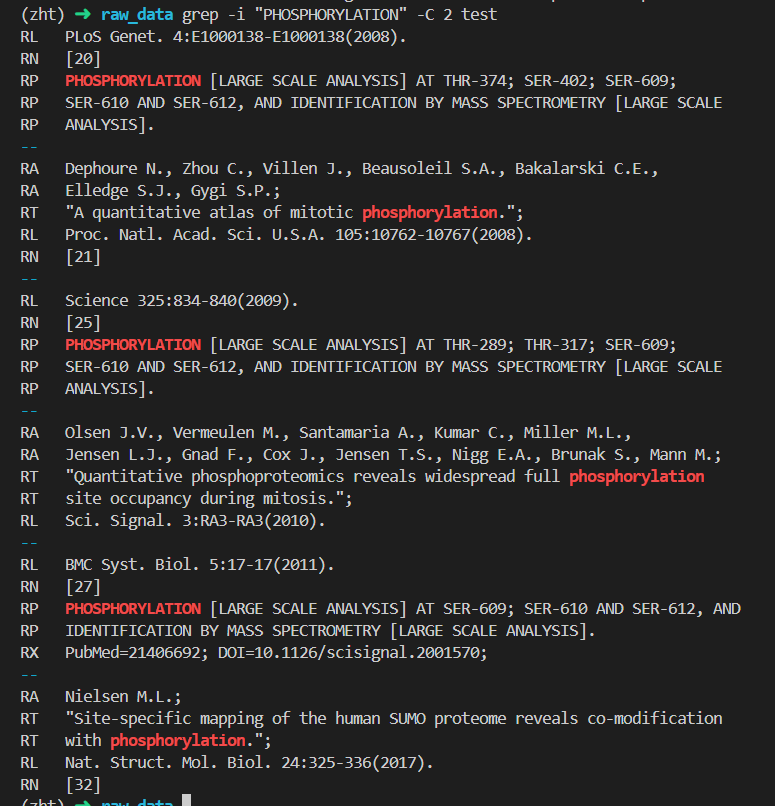
位点 289:Phosphothreonine x-25
位点 317:Phosphothreonine x-25
位点 317:Phosphothreonine x-25
位点 349:Sumoylated lysine (Bronze)
位点 374:Phosphothreonine x-20
位点 374:Phosphothreonine x-20
位点 402:Phosphoserine x-20
位点 402:Phosphoserine x-20
位点 407:Phosphotyrosine
位点 450:Phosphoserine
位点 461:Phosphoserine
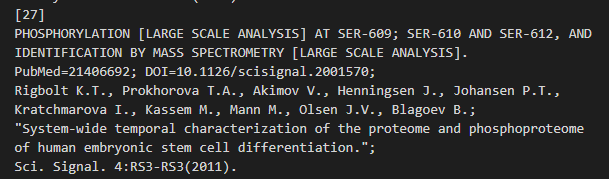
位点 609:Phosphoserine x-20 x-25 x-27
位点 610:Phosphoserine x-20 x-25 x-27
位点 612:Phosphoserine x-20 x-25 x-27
位点 689:Sumoylated lysine (Gold) x-29 x-31
位点 690:Sumoylated lysine (Bronze)
位点 717:Phosphothreonine
发现text中的PTM注释信息是不全面的,而且这个注释很奇怪,既不是modified residue里的,也不是large scale data里的——》text格式不能用
法2:通过下载的注释excel信息
如果是excel的表格的话:
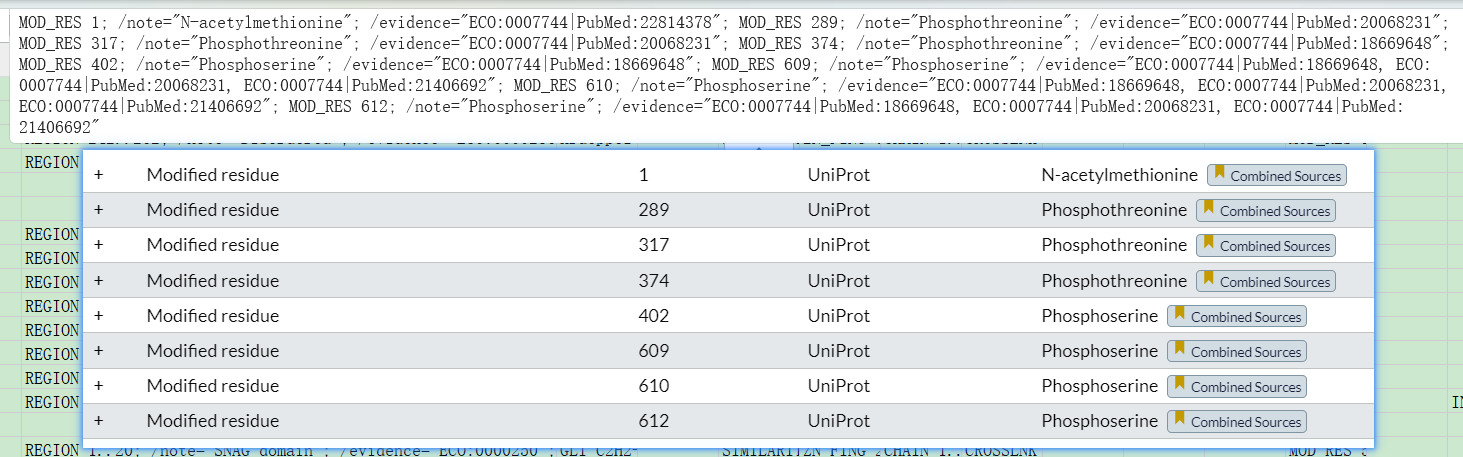
modified residue里的信息倒是全面的,但是large scale data的修饰注释就完全丢失了
下载gff文件:

可以发现还是8个,还是modified residue里的,但是就是没有large scale 的data
下载XML文件发现同样只能下载8个:

json文件倒是有
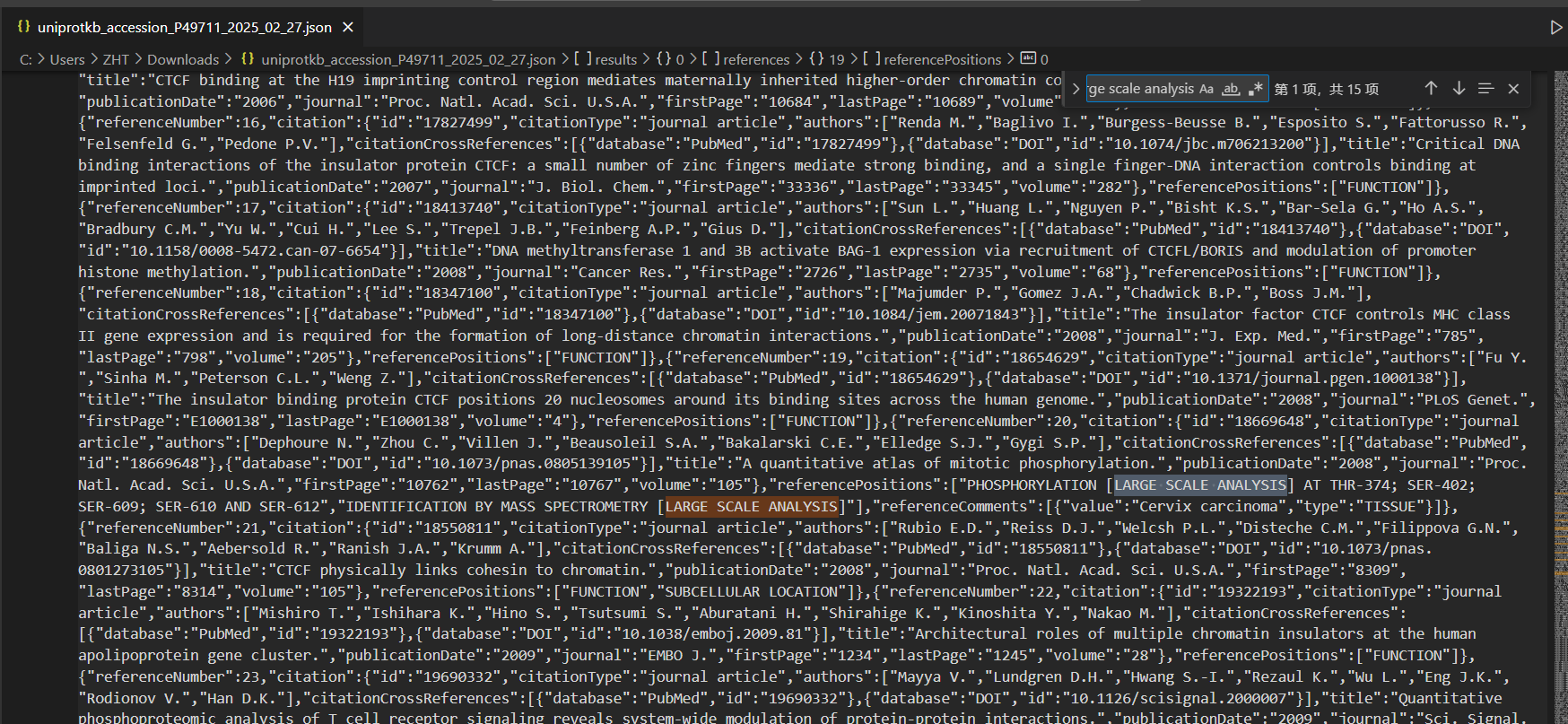
tsv文件的话同理,和excel一样,只有modified residue

法3:通过直接在网页下载json文件
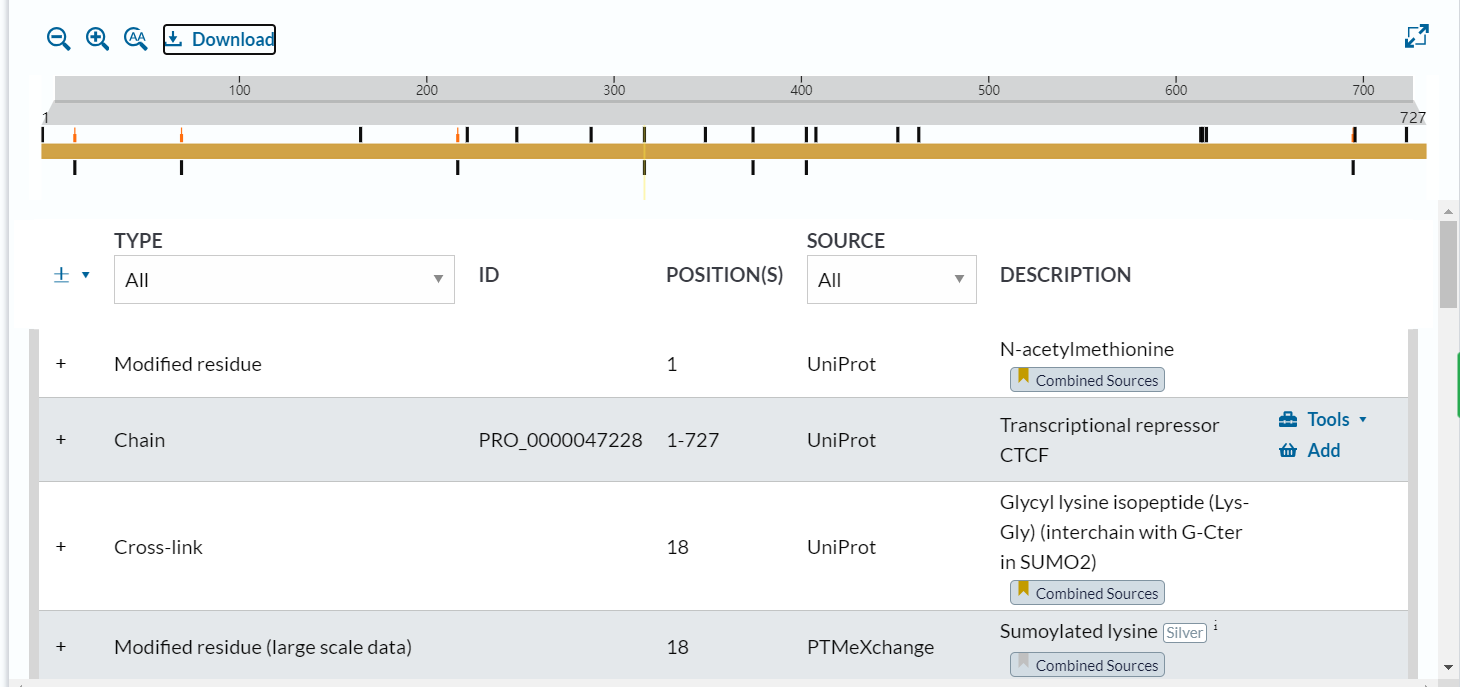

但这种方法还不如直接手动抄录,还是一个一个蛋白的,没法自动化
——》综上,如果要使用只有uniprot中的人工整理的注释的话,建议下载gff格式比较好
另外按理来说json格式是能够获取所有的注释的
2,利用python或R自动获取数据:
当然了,我们是学生信的,做计算的,如果不会自动获取大规模数据,也未免太丢人了。
https://www.biostars.org/p/261823/
https://www.biostars.org/p/197473/
(1)python使用bioservices
https://bioservices.readthedocs.io/en/main/
https://bioservices.readthedocs.io/en/main/references.html#module-bioservices.uniprot
github官网见https://github.com/cokelaer/bioservices


from bioservices import UniProt
u = UniProt(verbose=False)
data = u.search("zap70+and+taxonomy_id:9606", frmt="tsv", limit=3,
columns="id,length,accession, gene_names")
print(data)
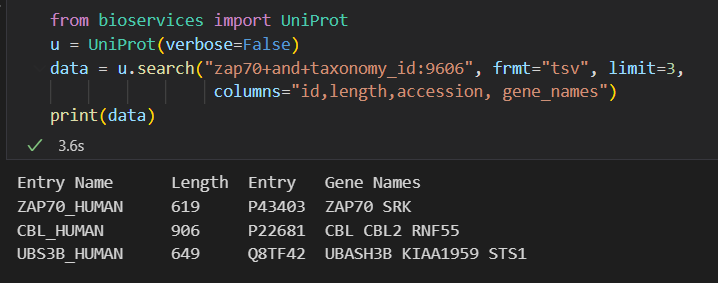
如果我们这样直接在uniprot中搜索的话:
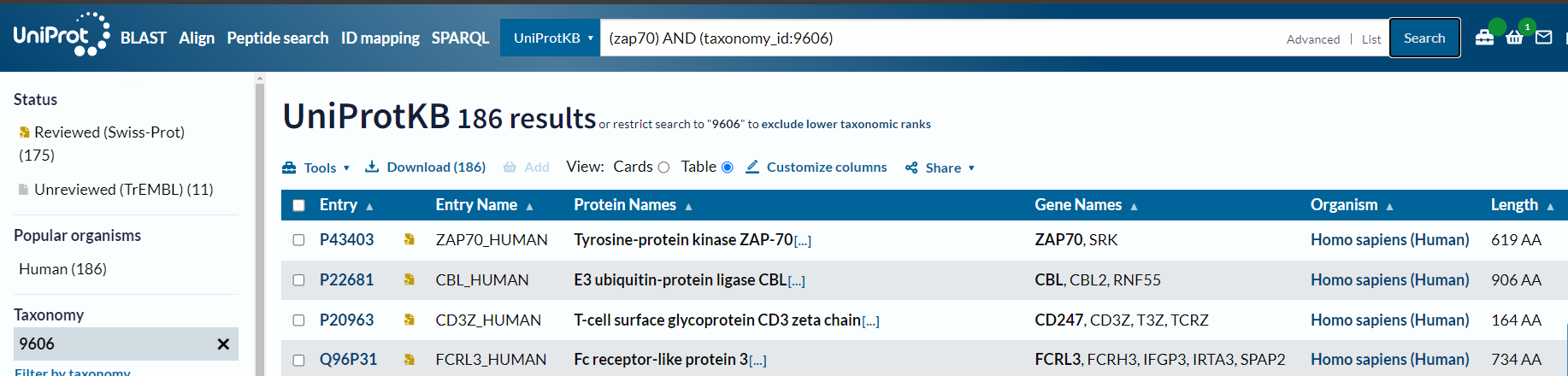
我们再来仔细研究一下这些参数:
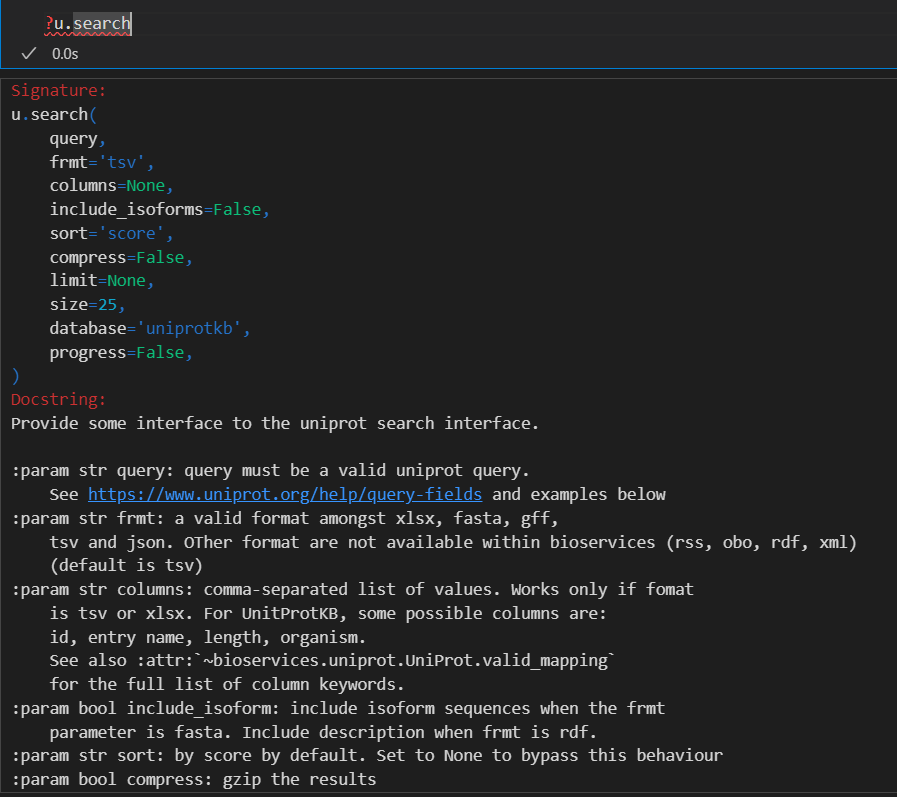
首先是query,可以参考:
https://www.uniprot.org/help/query-fields

我主要关注的是其他数据库的参考引用:
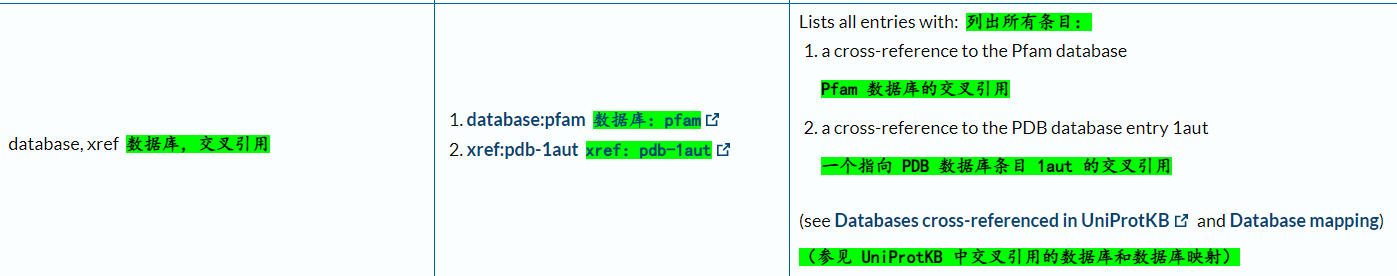
查看文档:
https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/docs/dbxref.txt

https://www.uniprot.org/help/id_mapping
这里其实能够满足uniprot中蛋白质id的mapping等工作



然后是frmt参数:其实就是数据格式,
和能够直接在uniprot结果界面中下载的格式一致

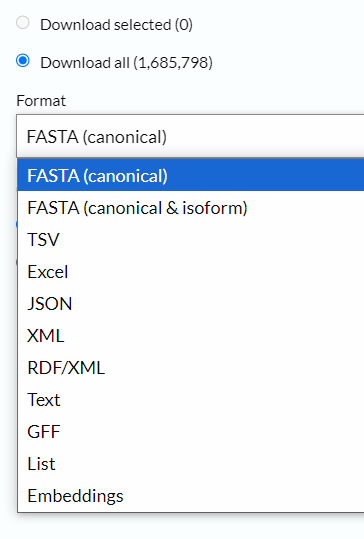
依据前面的经验,我们选择的下载格式是tsv、fasta以及gff格式
还有1个column参数:
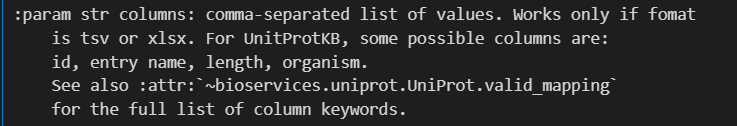
只有当下载的数据格式frmt是tsv或者是xlsx的时候,该参数才有效
但是我们可以看到,使用的列比较有限

当下载fasta序列格式的时候,我们一般不选择isoform,就是异构体的数据我们不要
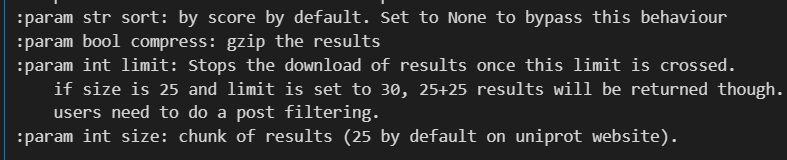
然后就是其他的参数,
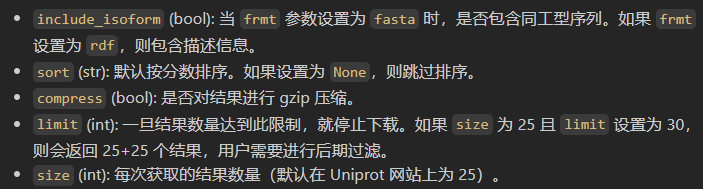
总之所有参数如下:
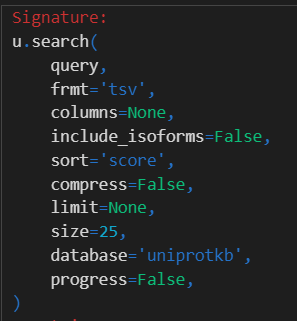
如果多个数据库参考
# reviewed
from bioservices import UniProt
u = UniProt(verbose=False)
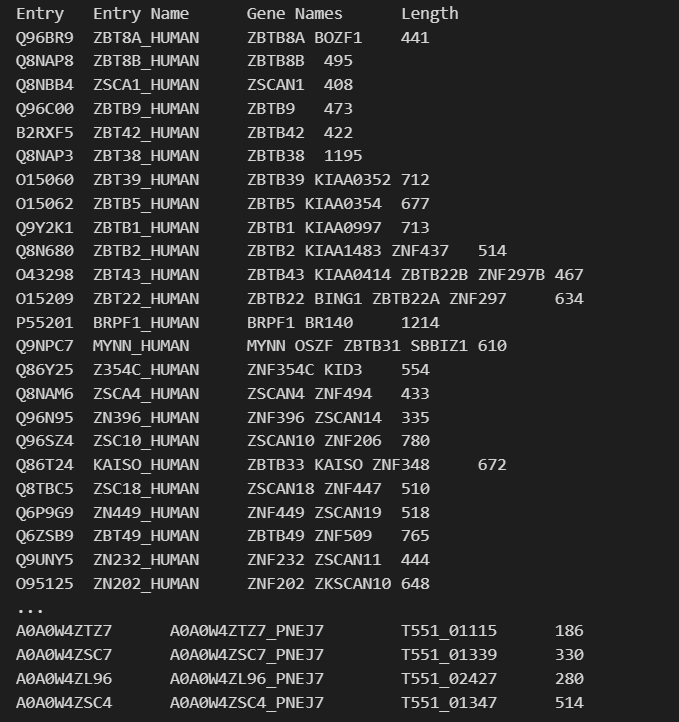
c2h2_zf_PROSITE = u.search("xref:PROSITE-PS00028+and+xref:PROSITE-PS50157+and+organism_name:Human", frmt="tsv",
columns="accession,id,gene_names,length")
print(c2h2_zf_PROSITE)
import io
import pandas as pd
# 将 TSV 格式的字符串转换为 DataFrame
df = pd.read_csv(io.StringIO(c2h2_zf_PROSITE), sep='\t')
# 现在你可以使用 df.shape 来获取 DataFrame 的形状
print(df.shape)
print(df)
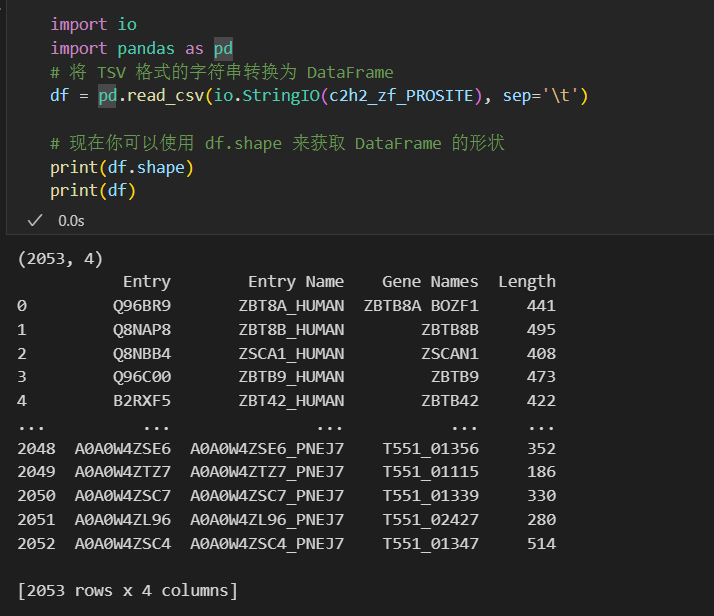
问题是实际上筛选的时候有2.5k左右
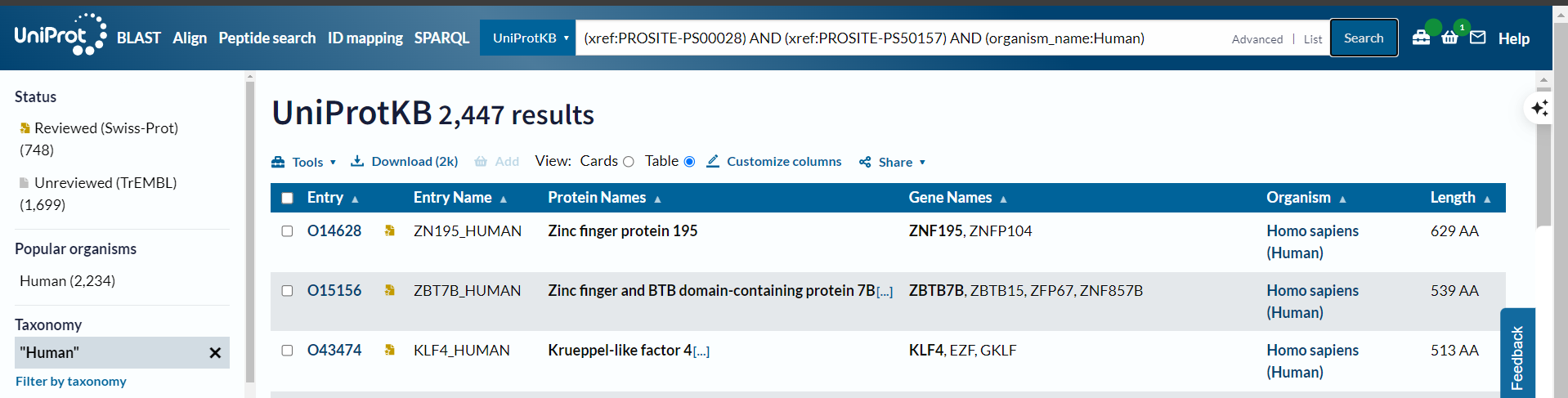
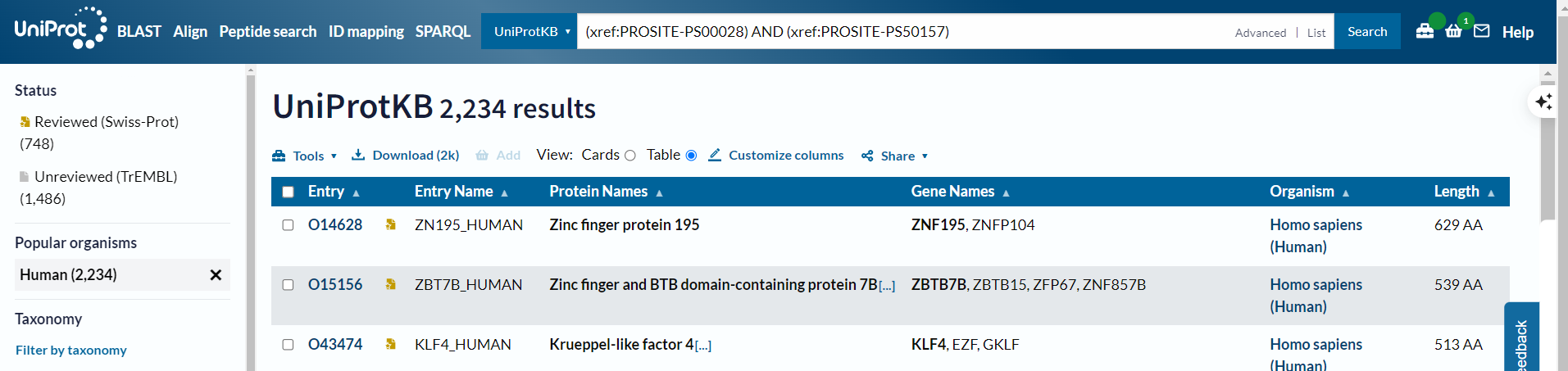
所以正确的方式不是使用organism_name,而是使用taxonomy_id:9606
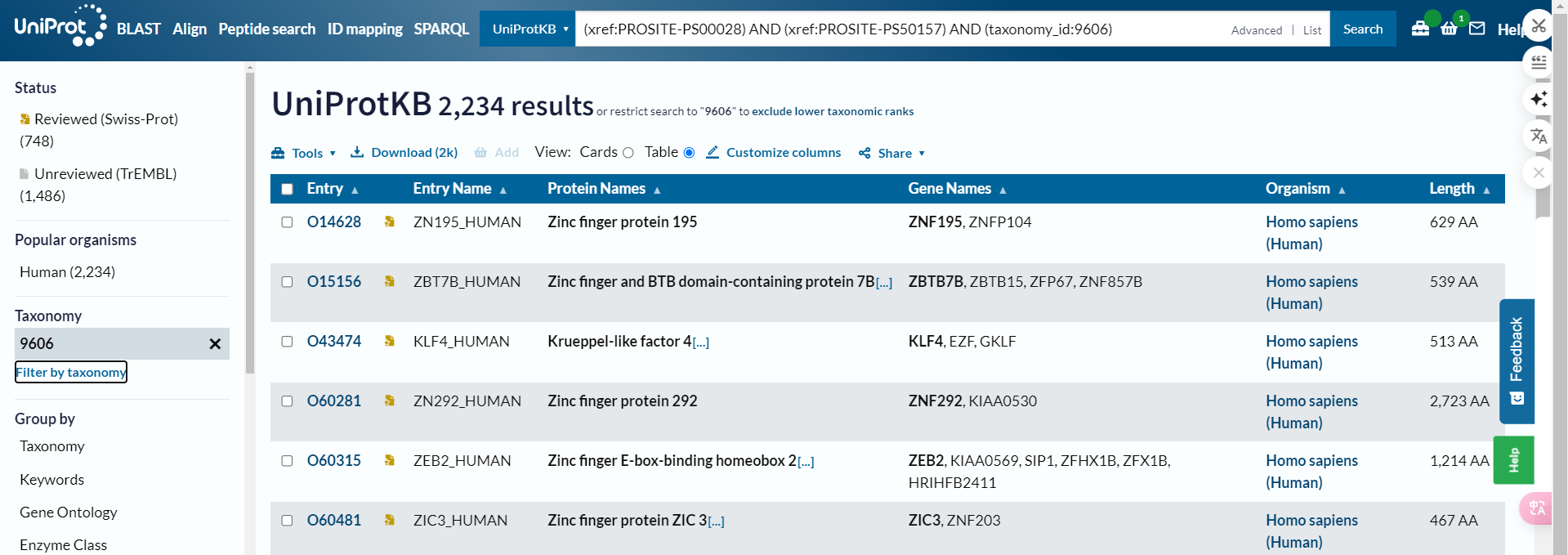
也就是说在uniprot中直接搜索的时候正确的检索方式是:
(xref:PROSITE-PS00028) AND (xref:PROSITE-PS50157) #在检索界面中选中human,√
(xref:PROSITE-PS00028) AND (xref:PROSITE-PS50157) AND (taxonomy_id:9606) #√
(xref:PROSITE-PS00028) AND (xref:PROSITE-PS50157) AND (organism_name:Homo sapiens) #也可以√
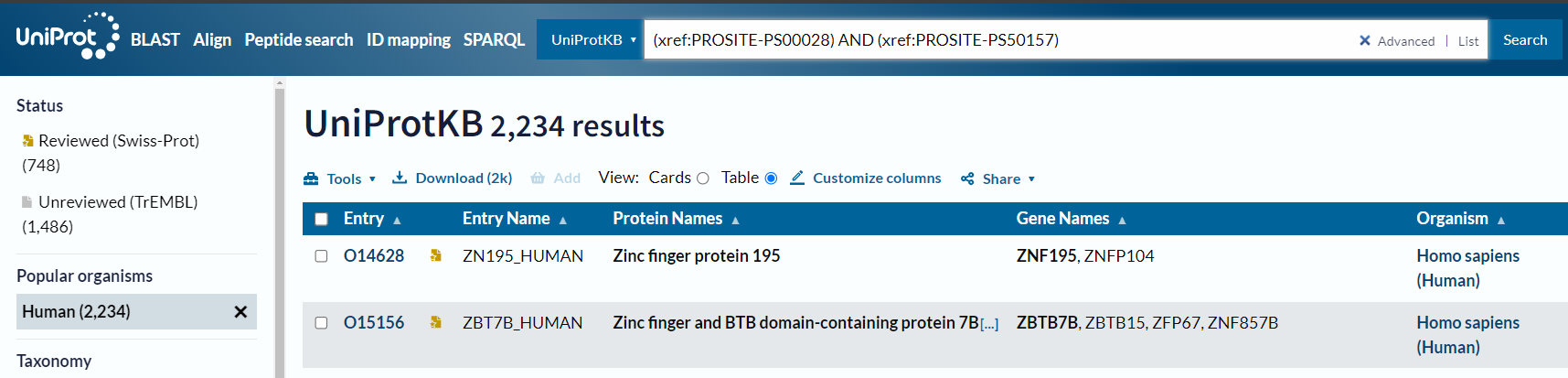
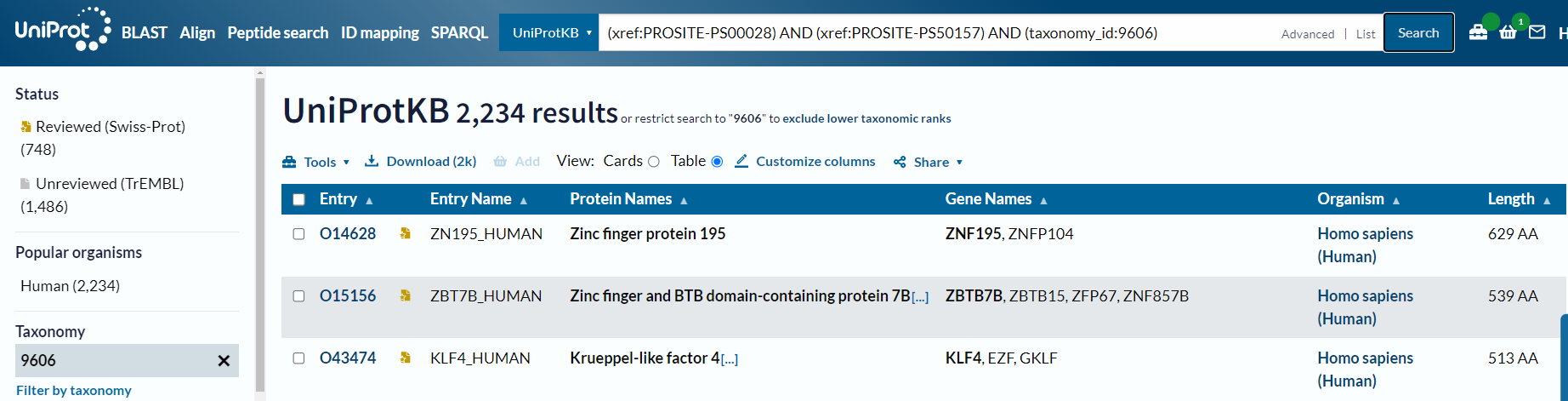
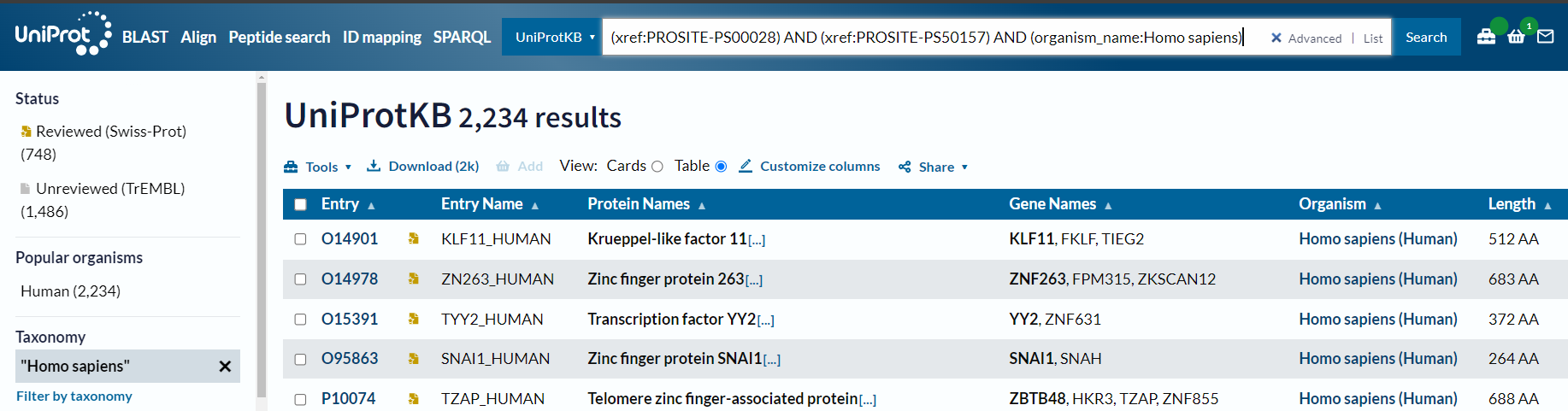
但是唯独human不行:
(xref:PROSITE-PS00028) AND (xref:PROSITE-PS50157) AND (organism_name:Human) # X
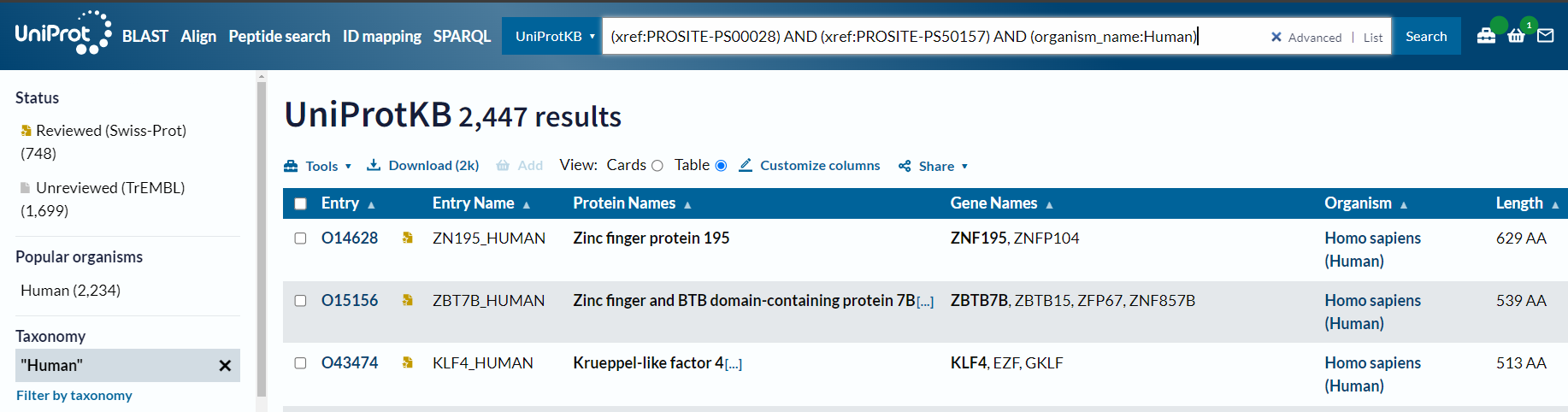
事实上我们可以从NCBI中获取该ID的意义,就是对应homo sapiens
https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=info&id=9606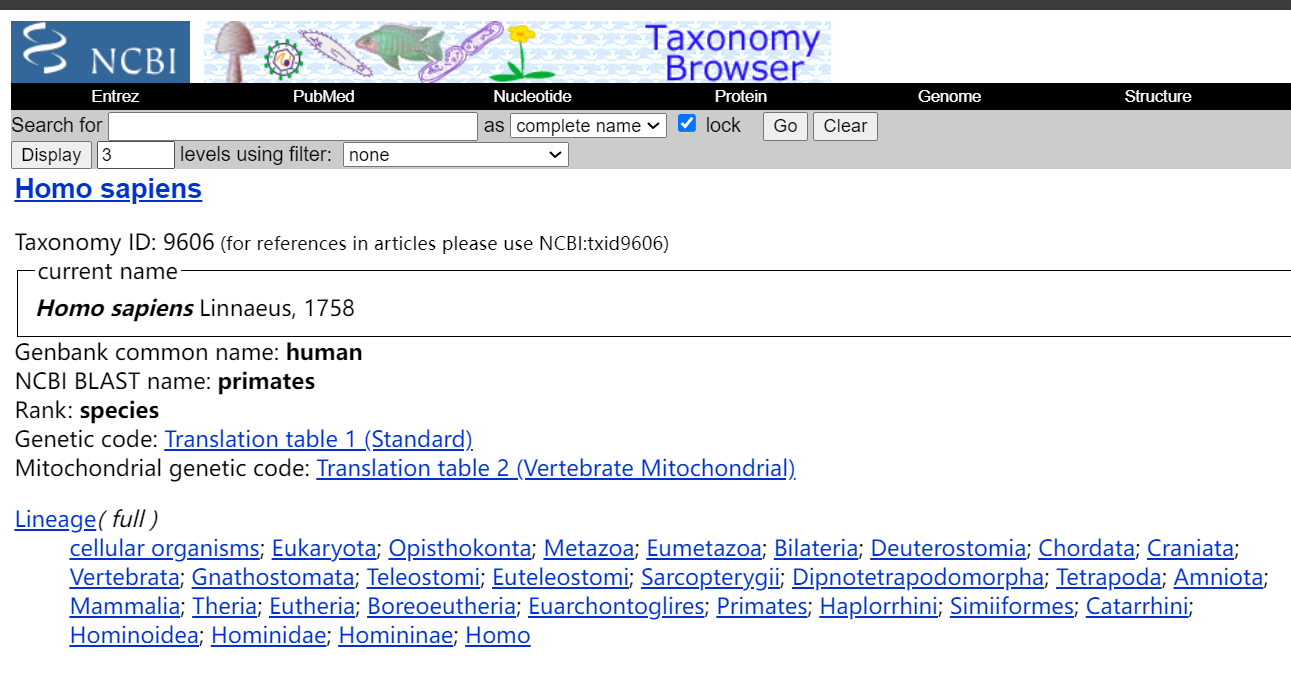
所以正确的使用方式是:
# reviewed
from bioservices import UniProt
u = UniProt(verbose=False)
c2h2_zf_PROSITE = u.search("xref:PROSITE-PS00028+and+xref:PROSITE-PS50157+and+taxonomy_id:9606", frmt="tsv",
columns="accession,id,gene_names,length")
print(c2h2_zf_PROSITE)
import io
import pandas as pd
# 将 TSV 格式的字符串转换为 DataFrame
df = pd.read_csv(io.StringIO(c2h2_zf_PROSITE), sep='\t')
# 现在你可以使用 df.shape 来获取 DataFrame 的形状
print(df.shape)
print(df)
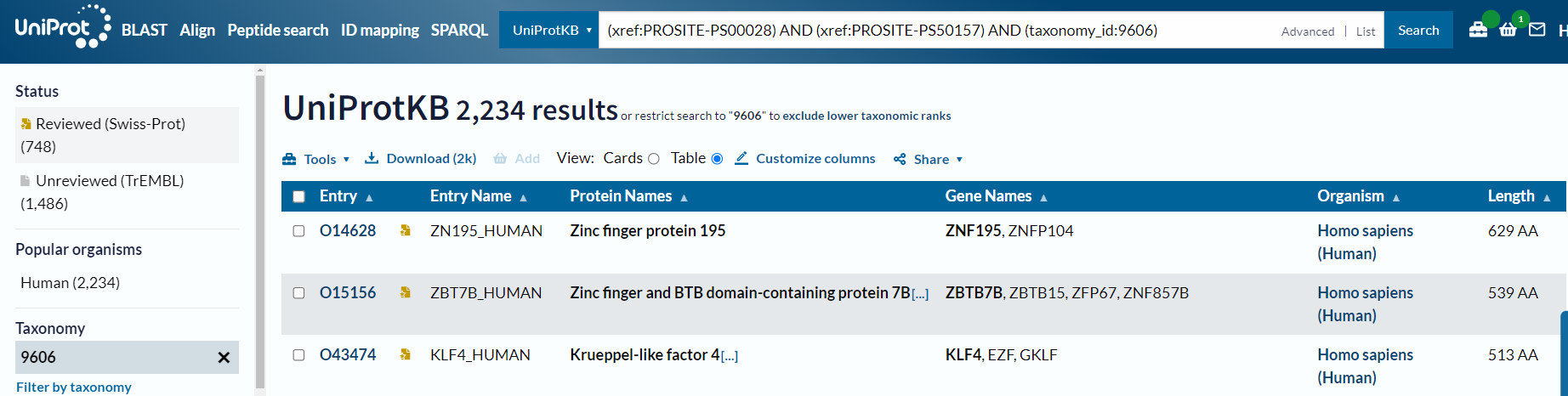
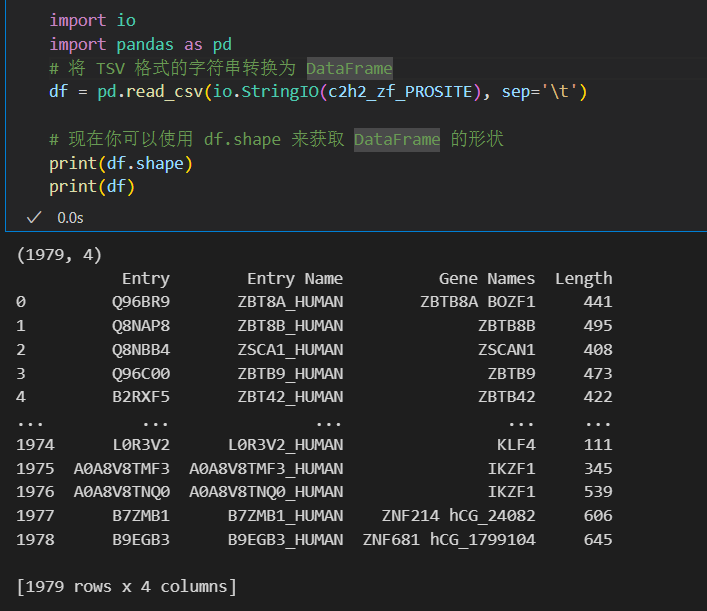
只能说数据上确实还是差点
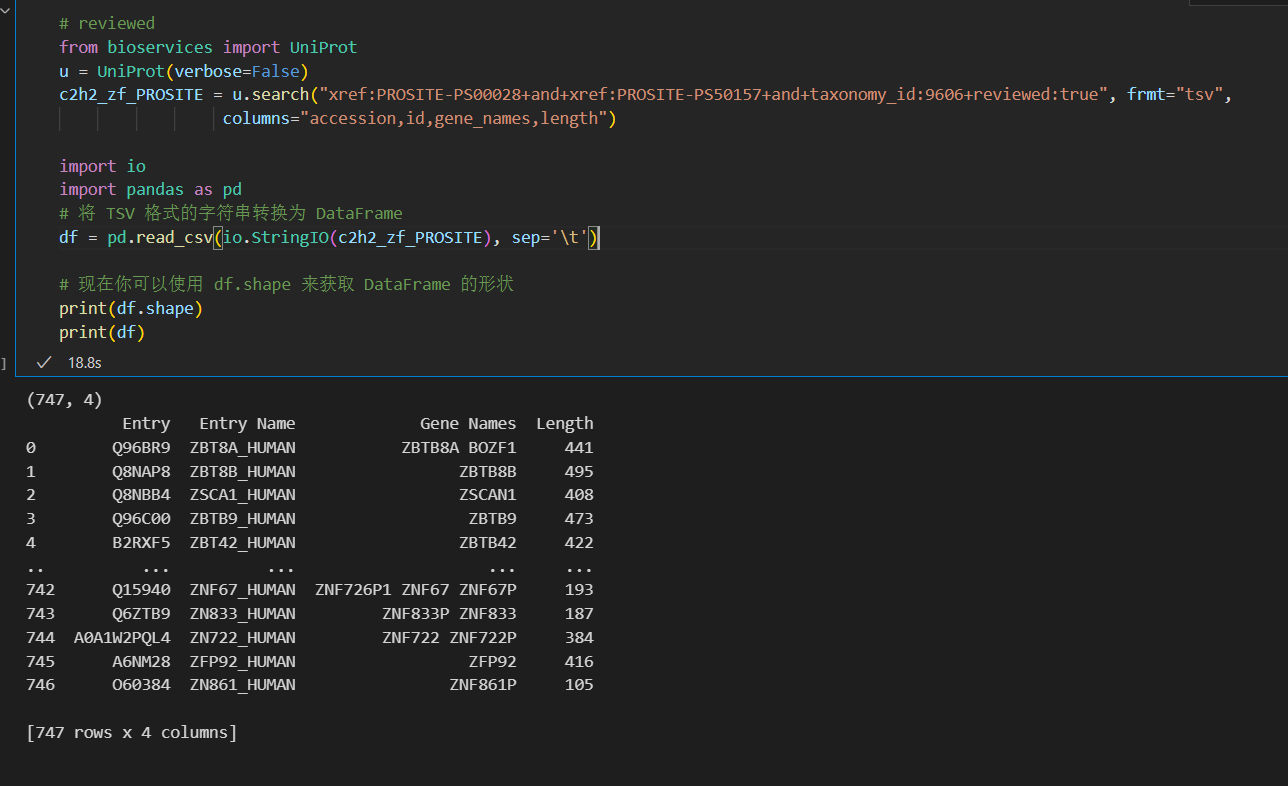
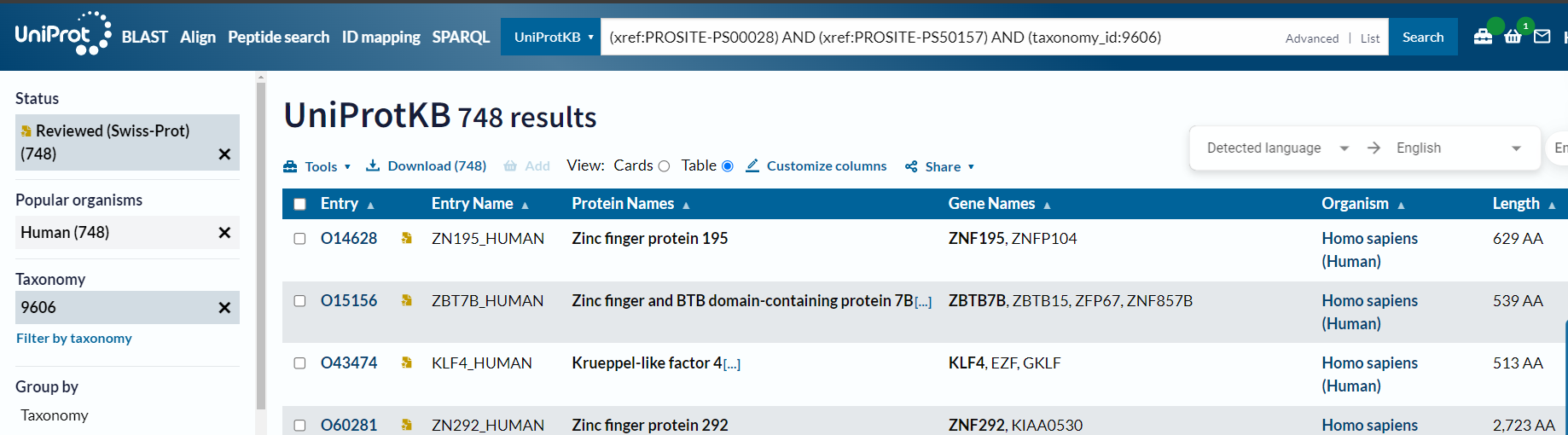
但是奇了怪了,还是差了一个id
——》总之,使用该python库
https://bioservices.readthedocs.io/en/main/protein.html
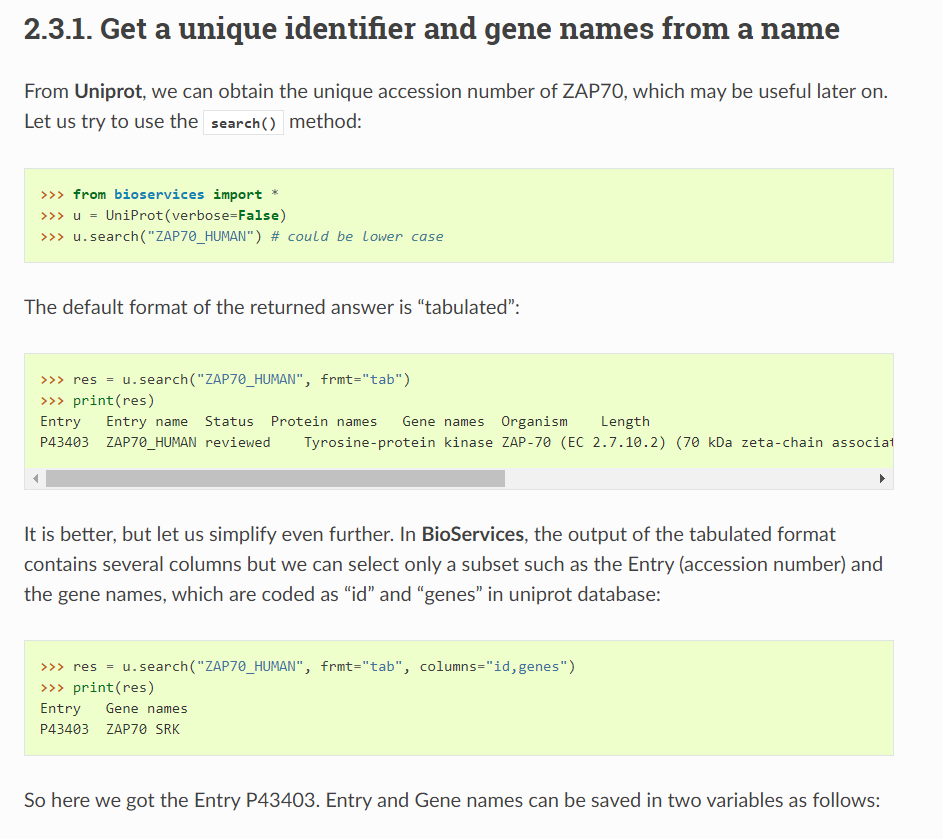
——》但是获取的数据信息实在是有限,尤其是如果我们想获取更多的细节信息的时候,比如说domain、structure或者是PTM等每个蛋白质条目点进去才能获取的信息的时候
https://stackoverflow.com/questions/33689703/ptm-data-from-bioservices-uniprot
https://stackoverflow.com/questions/33689703/ptm-data-from-bioservices-uniprot
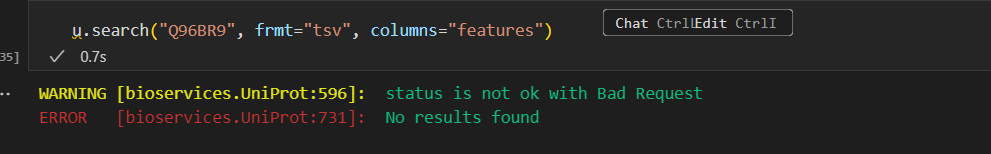

但是很奇怪,不管用,也许是version问题
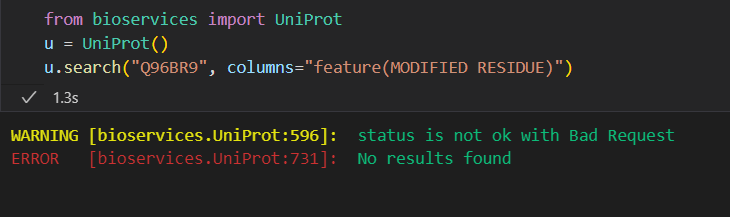
事实上我们正常pip之后的操作是
pip install bioservices
# 查看版本信息
pip show bioservices
pip install --upgrade bioservices
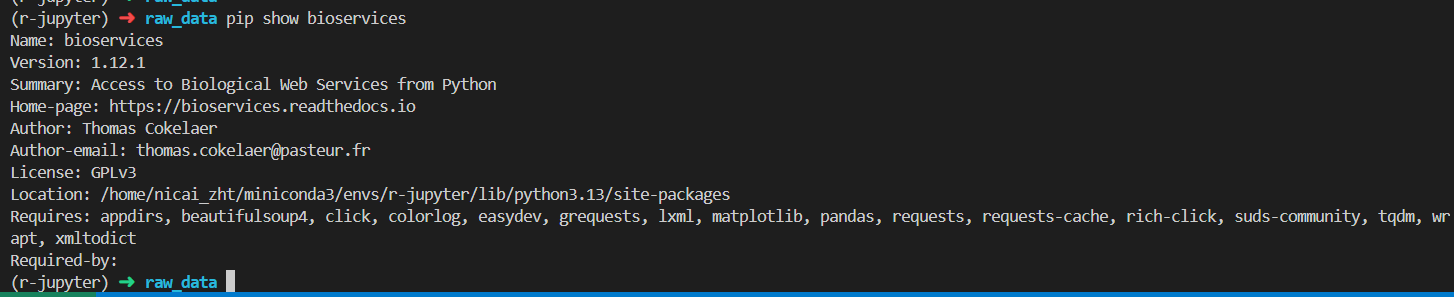
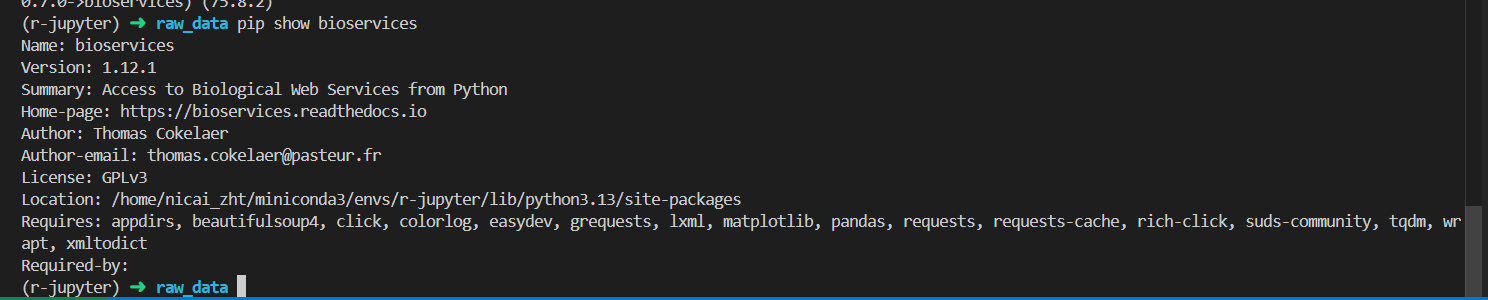
已经是最新版本了,所以该库实际上并没有解决我们的问题
import io
import pandas as pd
from collections import Counter
from bioservices import UniProt
u = UniProt()
n_results = []
for i in range(100):
res = u.search("P42328", frmt="tab", columns="id, sequence")
# Store results in pandas dataframe
df_res = pd.read_csv(io.StringIO(res), sep="\t")
n_results.append(len(df_res))
print(Counter(n_results))
事实上我们看看对于uniprot中一些feature,是怎么引用的:
https://github.com/cokelaer/bioservices/issues/163
https://github.com/cokelaer/bioservices/issues/237
从https://github.com/cokelaer/bioservices/issues/147中我们可以看到有get_df 函数
我们也使用了这个函数,但是发现实际上是有110列的,所以我们的数据是能够从这里获取的:
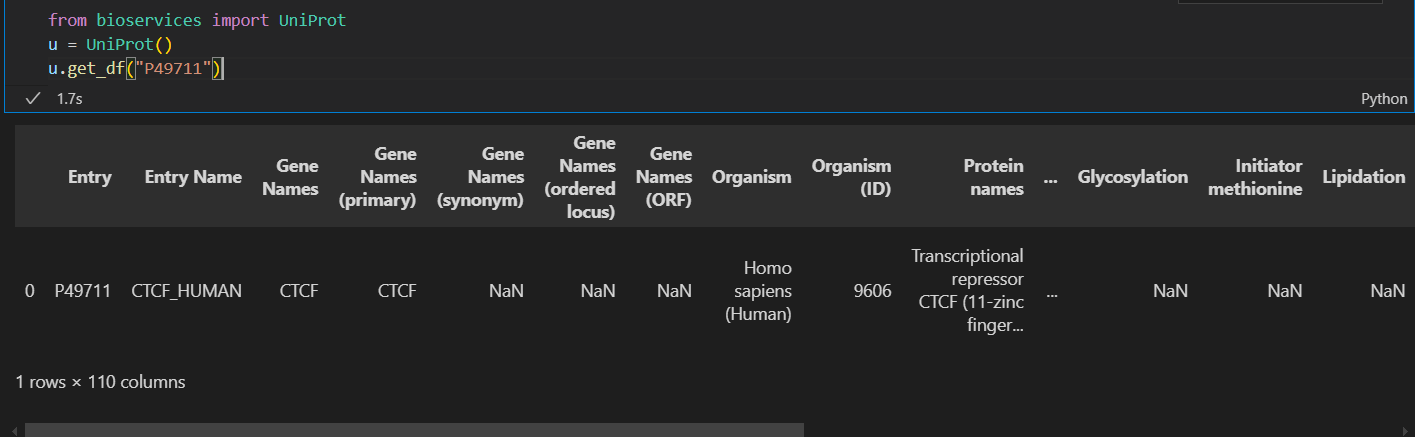
我们需要查看所有列的名字
import pandas as pd
from bioservices import UniProt
u = UniProt()
u.get_df("P49711")
print(u.get_df("P49711").columns)
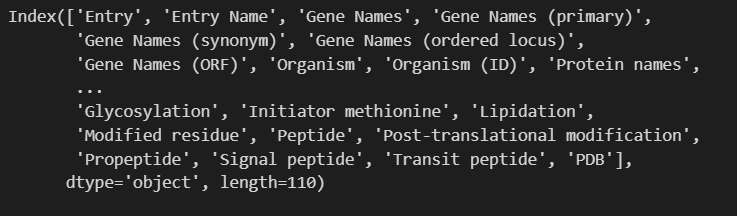
因为获取的是index类型的数据
所以我们如果要查看的话需要将数据类型进行转换:可以使用tolist转换为列表
import pandas as pd
from bioservices import UniProt
u = UniProt()
u.get_df("P49711")
print(u.get_df("P49711").columns)
print(u.get_df("P49711").columns.tolist())
'Entry', 'Entry Name', 'Gene Names', 'Gene Names (primary)', 'Gene Names (synonym)', 'Gene Names (ordered locus)', 'Gene Names (ORF)', 'Organism', 'Organism (ID)', 'Protein names', 'Proteomes', 'Taxonomic lineage', 'Virus hosts', 'Fragment', 'Sequence', 'Length', 'Mass', 'Gene encoded by', 'Alternative products (isoforms)', 'Erroneous gene model prediction', 'Mass spectrometry', 'Polymorphism', 'RNA Editing', 'Sequence caution', 'Alternative sequence', 'Natural variant', 'Non-adjacent residues', 'Non-standard residue', 'Non-terminal residue', 'Sequence conflict', 'Sequence uncertainty', 'Sequence version', 'Coiled coil', 'Compositional bias', 'Domain [CC]', 'Domain [FT]', 'Motif', 'Protein families', 'Region', 'Repeat', 'Zinc finger', 'Absorption', 'Active site', 'Activity regulation', 'Binding site', 'Catalytic activity', 'Cofactor', 'DNA binding', 'EC number', 'Function [CC]', 'Kinetics', 'Pathway', 'pH dependence', 'Redox potential', 'Site', 'Temperature dependence', 'Gene Ontology (GO)', 'Gene Ontology (biological process)', 'Gene Ontology (molecular function)', 'Gene Ontology (cellular component)', 'Gene Ontology IDs', 'Interacts with', 'Subunit structure', 'Developmental stage', 'Induction', 'Tissue specificity', 'PubMed ID', 'Date of creation', 'Date of last modification', 'Date of last sequence modification', 'Entry version', '3D', 'Beta strand', 'Helix', 'Turn', 'Subcellular location [CC]', 'Intramembrane', 'Topological domain', 'Transmembrane', 'Annotation', 'Caution', 'Comments', 'Features', 'Keywords', 'Keyword ID', 'Miscellaneous [CC]', 'Protein existence', 'Tools', 'Reviewed', 'UniParc', 'Allergenic Properties', 'Biotechnological use', 'Disruption phenotype', 'Involvement in disease', 'Mutagenesis', 'Pharmaceutical use', 'Toxic dose', 'Chain', 'Cross-link', 'Disulfide bond', 'Glycosylation', 'Initiator methionine', 'Lipidation', 'Modified residue', 'Peptide', 'Post-translational modification', 'Propeptide', 'Signal peptide', 'Transit peptide', 'PDB'
最好是转换为dataframe的格式,但是df展示的时候只能最大展示20列,所有会有省略号
我们可以设置一下
此处我们需要设置max_rows这个参数,在设置前后恢复为60
# 显示所有列
pd.set_option('display.max_columns',None)
# 显示所有行
pd.set_option('display.max_rows',None)
import pandas as pd
from bioservices import UniProt
u = UniProt()
u.get_df("P49711")
# 是index类型
print(u.get_df("P49711").columns)
# 可以转换为list
print(u.get_df("P49711").columns.tolist())
# 设置显示的最大行数为 None,以显示所有列
pd.options.display.max_rows = None
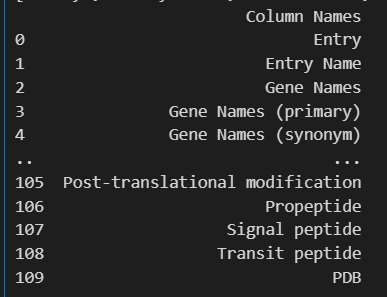
# 将列名转换为 DataFrame
columns_df = pd.DataFrame(u.get_df("P49711").columns, columns=['Column Names'])
columns_df
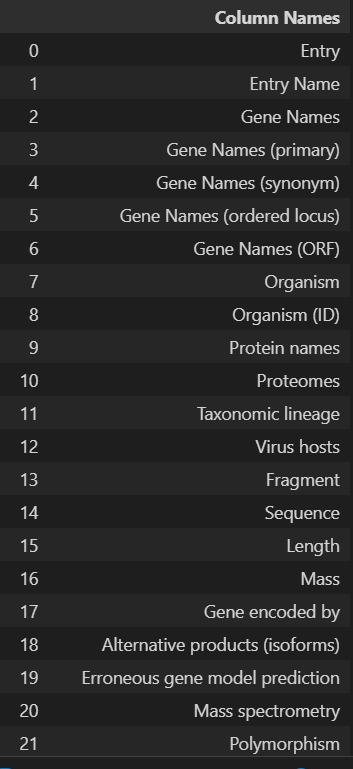
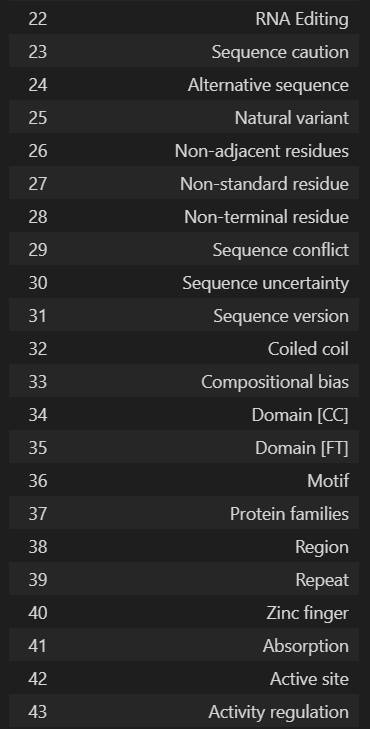

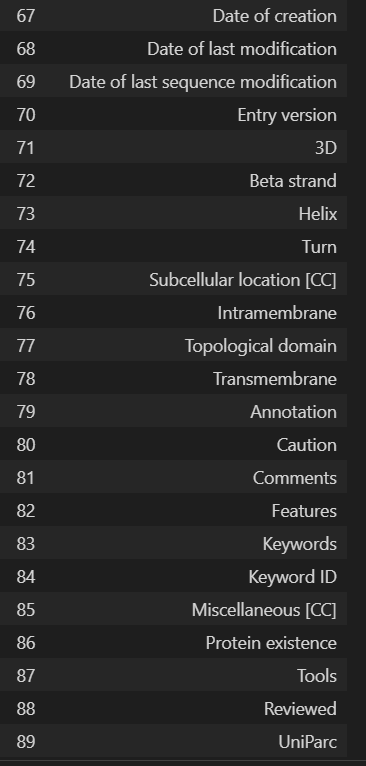
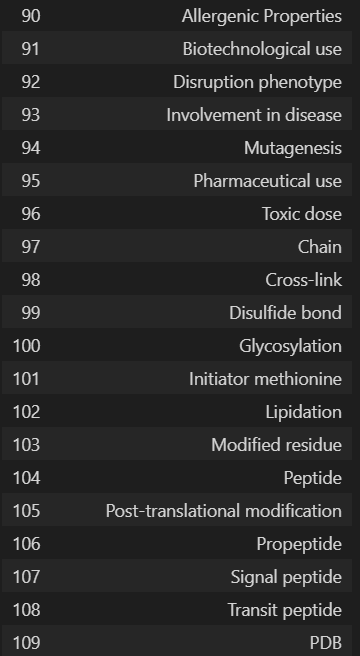
我们需要的列名是:
Entry
Entry Name
Organism
Organism (ID)
Protein names
Sequence
Domain [CC]
Domain [FT]
Motif
Zinc finger
Features
Reviewed
Modified residue
Post-translational modification
我们可以查看一下这几列:
# 选择特定的列
selected_columns = [
'Entry', 'Entry Name', 'Organism', 'Organism (ID)', 'Protein names',
'Sequence', 'Domain [CC]', 'Domain [FT]', 'Motif', 'Zinc finger',
'Features', 'Reviewed', 'Modified residue', 'Post-translational modification'
]
u.get_df("P49711")[selected_columns]
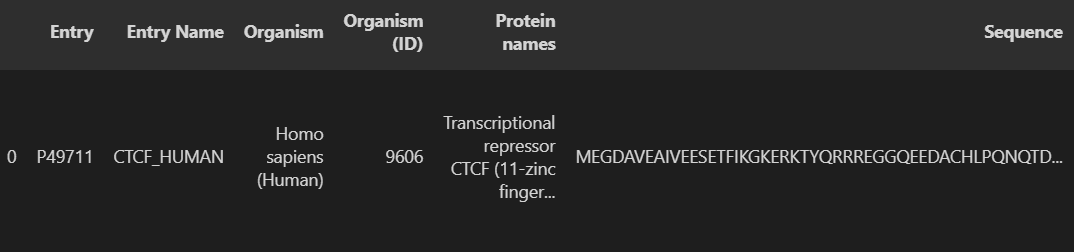
这里展示行或者列是无法看清楚具体的全文内容的,所以还是重定向到一个文本文件中再查看
u.get_df("P49711")["Modified residue"]
u = UniProt()
df = u.get_df("P49711")
# 将 'Modified residue' 列的内容写入一个文本文件
with open('modified_residue.txt', 'w') as file:
file.write(df["Modified residue"].to_string(index=False))
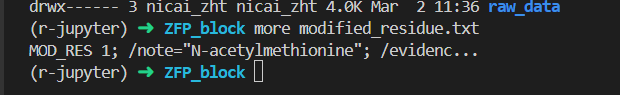
还是不行
再查看其他的github问题,应该是要重新使用search进行查看
https://github.com/cokelaer/bioservices/issues/122
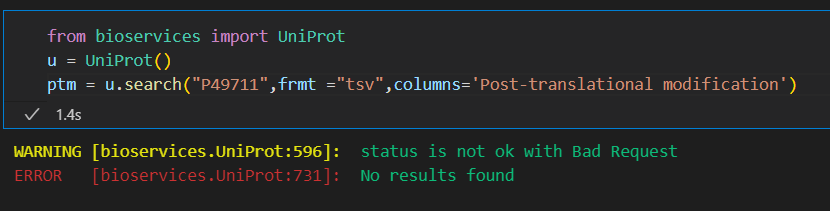
但是奇怪的是,还是不行:
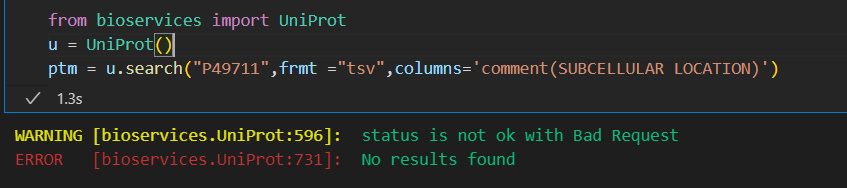
还是有问题,应该是网络问题
https://github.com/cokelaer/bioservices/issues/219
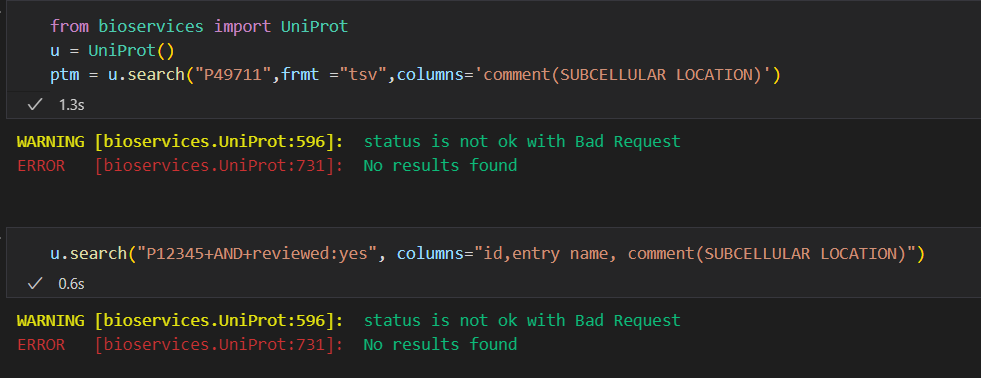
https://github.com/cokelaer/bioservices/issues/122
https://github.com/cokelaer/bioservices/issues/109
基本上所有在github中提供的解法在我这里都没有用,可能是因为这个工具出问题了,或者是最近uniprot的API又更改了
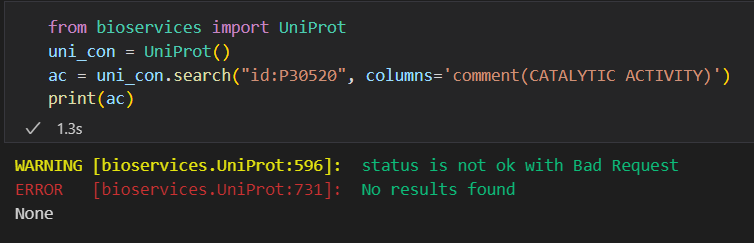
后来查看了一下,这个版本太新了:
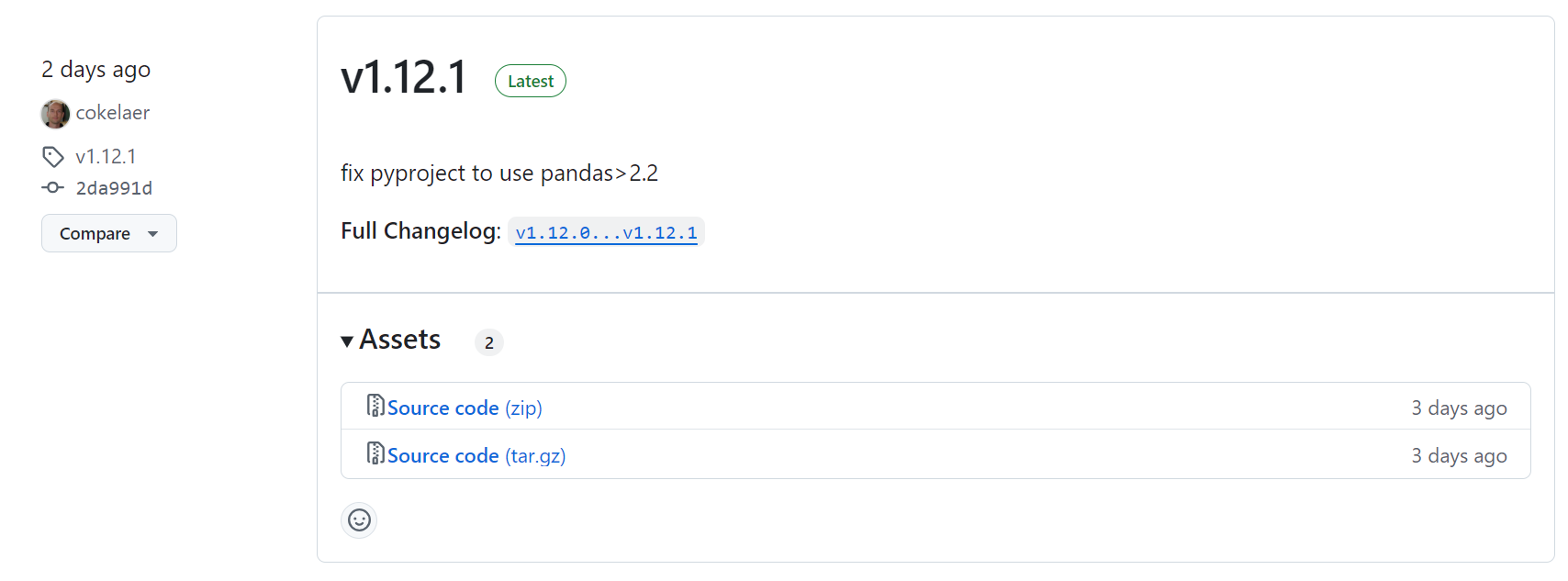
是2天前更新的,
也许我应该降级到一个稳定的版本试试看
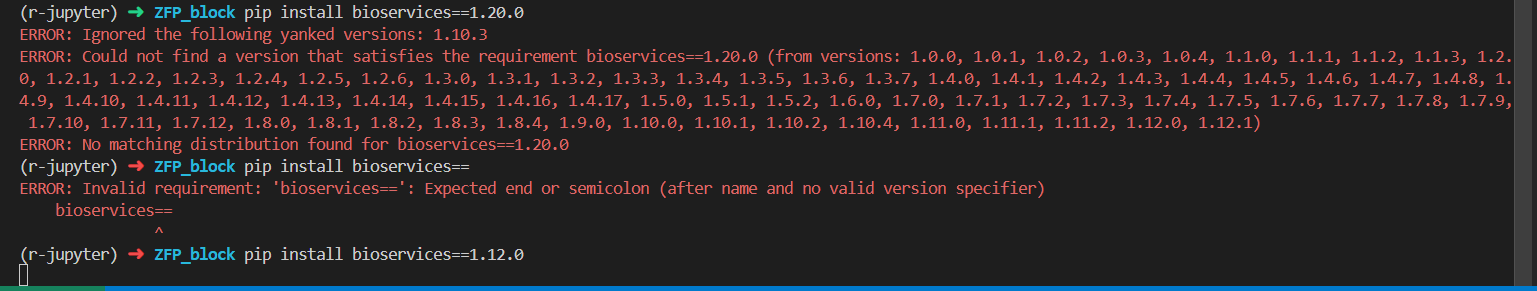
pip uninstall bioservices
pip install bioservices=1.12.0
有问题
还是用conda安装:
还是会有问题
——》此方法暂时搁置
还有其他的python工具,例如bio2bel-uniprot
https://mp.weixin.qq.com/s/VH7zTodBYf5KzHqnnLZavQ
(2)python+使用uniprot的API接口
查看官网中的说明:
https://www.uniprot.org/help/mod_res_large_scale

我们可以看到所需要的实际API是:
https://www.ebi.ac.uk/proteins/api/doc/#proteomicsApi
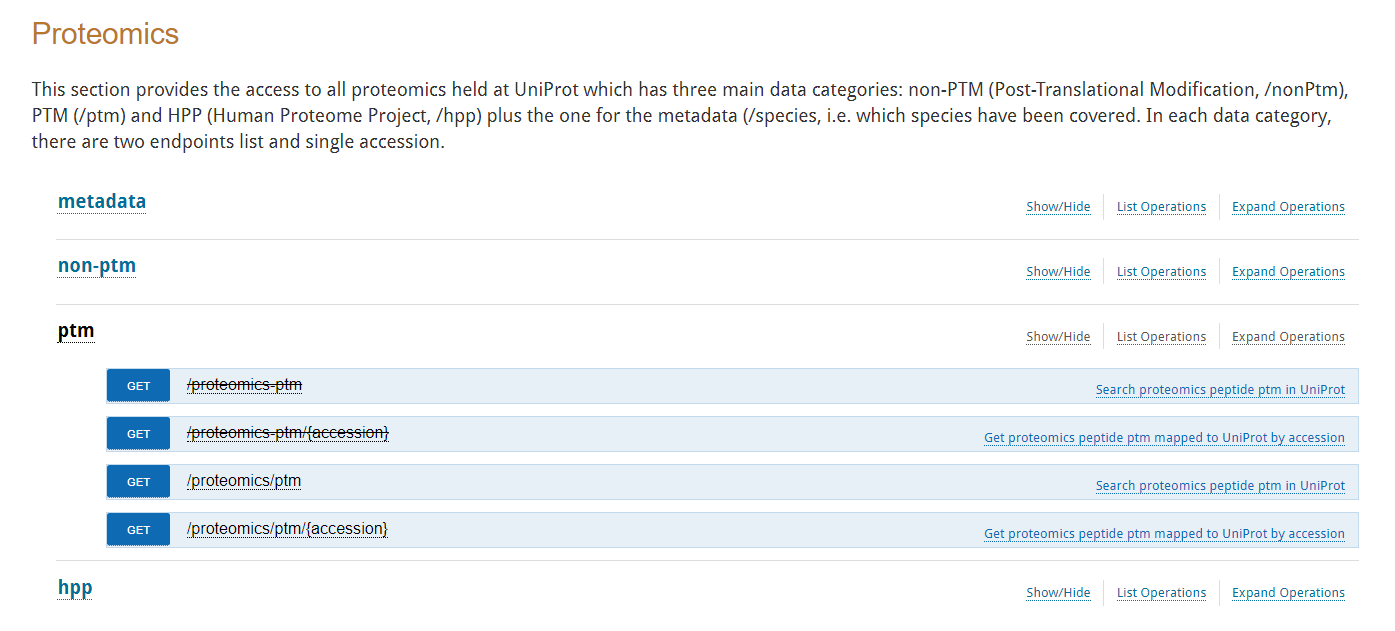
使用此处的PTM条目进行使用——》暂时搁置,具体如何使用该API的方法
https://stackoverflow.com/questions/33689703/ptm-data-from-bioservices-uniprot
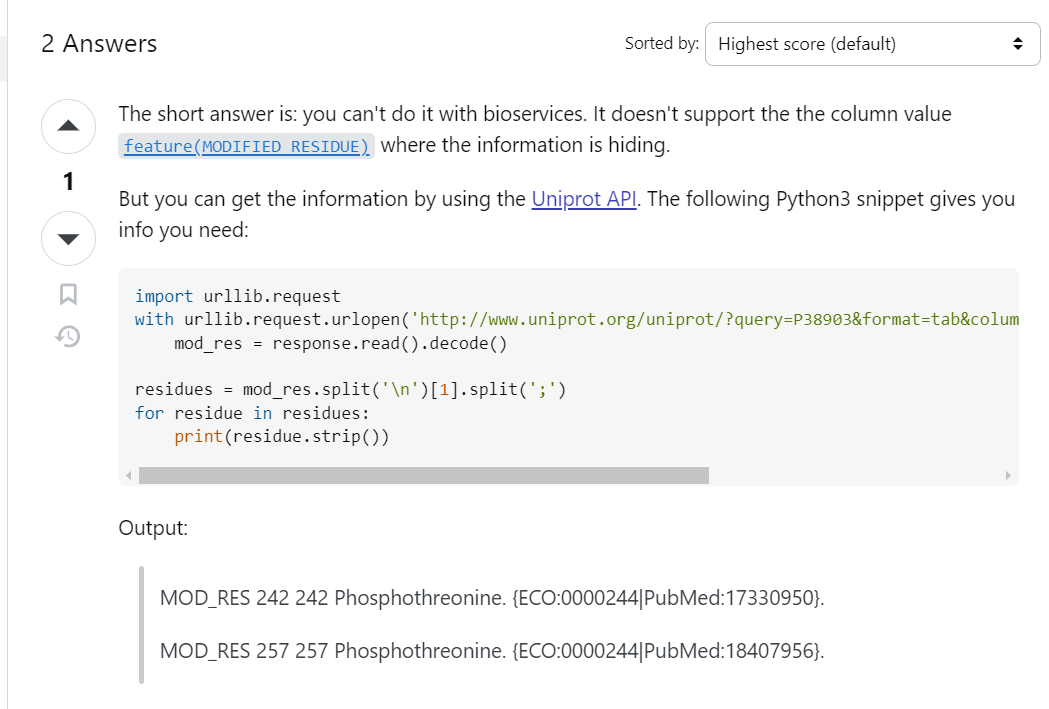
实际上还是不行
(3)uniprot+R:
如果使用python解决不了问题,可以使用R来处理
如果是R处理的话主要是2个工具:UniprotR以及rbioapi
https://github.com/moosa-r/rbioapi
https://github.com/Proteomicslab57357/UniprotR
https://github.com/Proteomicslab57357/UniprotR/blob/master/man/GetPTM_Processing.Rd
https://github.com/moosa-r/rbioapi/blob/master/vignettes/rbioapi_uniprot.Rmd
——》
我们先试试UniprotR这个R包:
直接在R中install有点麻烦,而且还是会有问题,还是使用conda
mamba install -y r-uniprotr=2.4.0
library(UniprotR)
??UniprotR
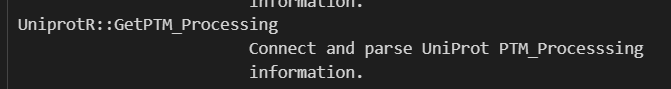
help(GetPTM_Processing)
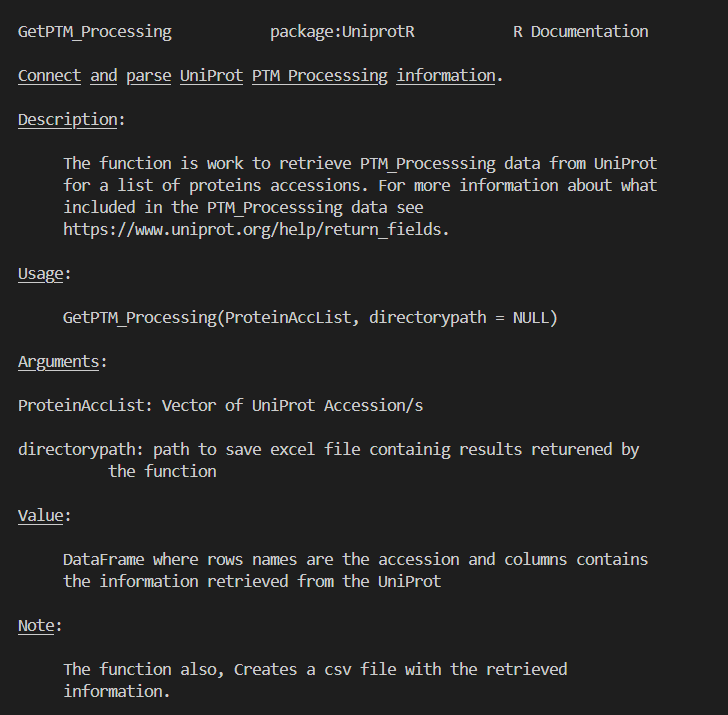
返回的值我们可以通过:
https://www.uniprot.org/help/return_fields
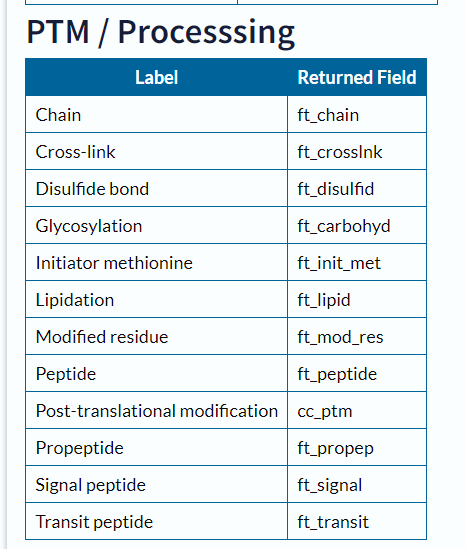
https://mp.weixin.qq.com/s/p4MGo-NX5bMu92NNpGwUlA
https://mp.weixin.qq.com/s/9JkQUijUx7r5UY4Q_eOa1Q
但是经常会有网络连接问题——》暂时搁置
(4)uniprot+rbioapi

这个工具还是挺新的,最近3年了
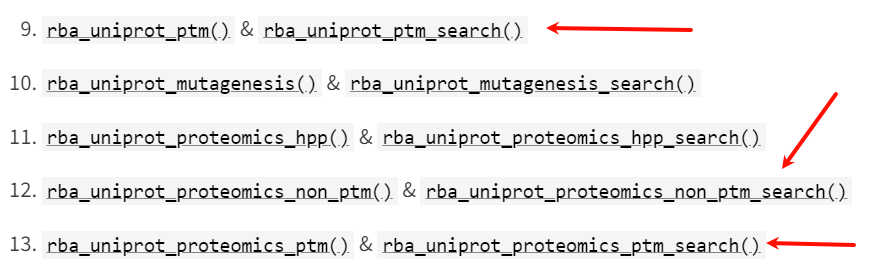
rba_uniprot_features_search(
accession = "P49711",
reviewed = TRUE,
organism = "Human",
categories = "PTM"
)
# Homo sapiens
# "MOD_RES"
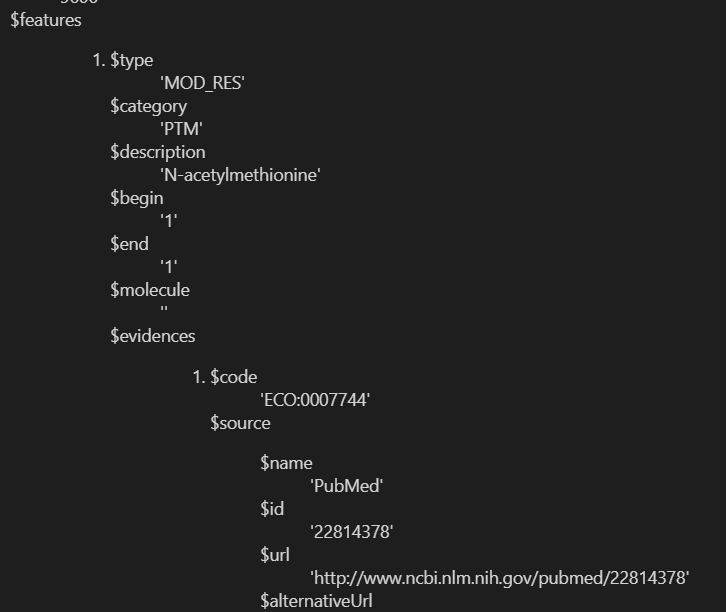
但是很不幸,这个函数也只能提供modified residue以及cross link的数据,不能提供large scale data的数据
# 使用R包rbioapi获取蛋白质的序列信息
# 参考https://rbioapi.moosa-r.com/articles/rbioapi_uniprot.html
# install.packages("remotes")
# remotes::install_github("moosa-r/rbioapi")
library(package = "rbioapi")
# 使用entry参数获取蛋白质的序列信息
ctcf_human <- rba_uniprot_proteins(accession = "P49711")
str(ctcf_human, 1)
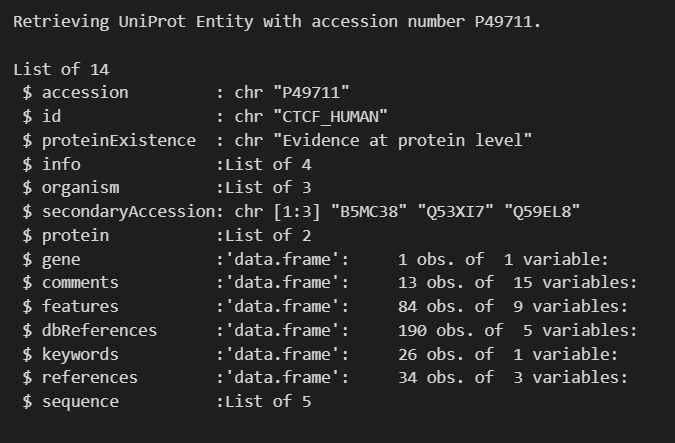
再回过头来看看我们的数据:
说明还是挺新的,更新也考虑到了
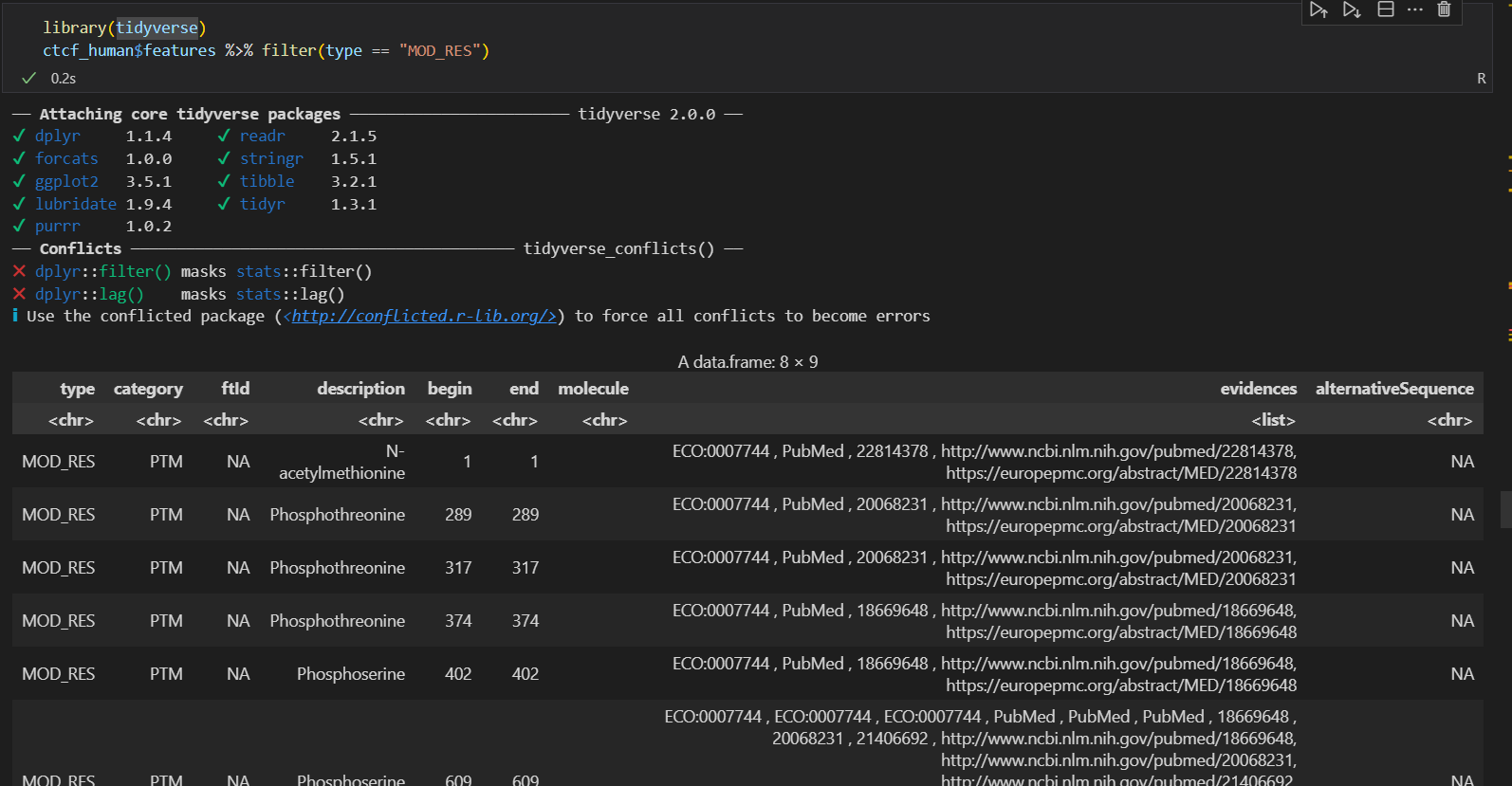
这个函数接口只能看到mod res的8种修饰
主要使用的函数还是:


其他几个都不建议使用,

主要使用rba_uniprot_proteomics_ptm
查证下来,只有13的函数还有用:
https://rbioapi.moosa-r.com/reference/rba_uniprot_proteomics_ptm.html
https://rbioapi.moosa-r.com/reference/rba_uniprot_proteomics_ptm_search.html
先来看看我们的函数:
所得到的结果的结构
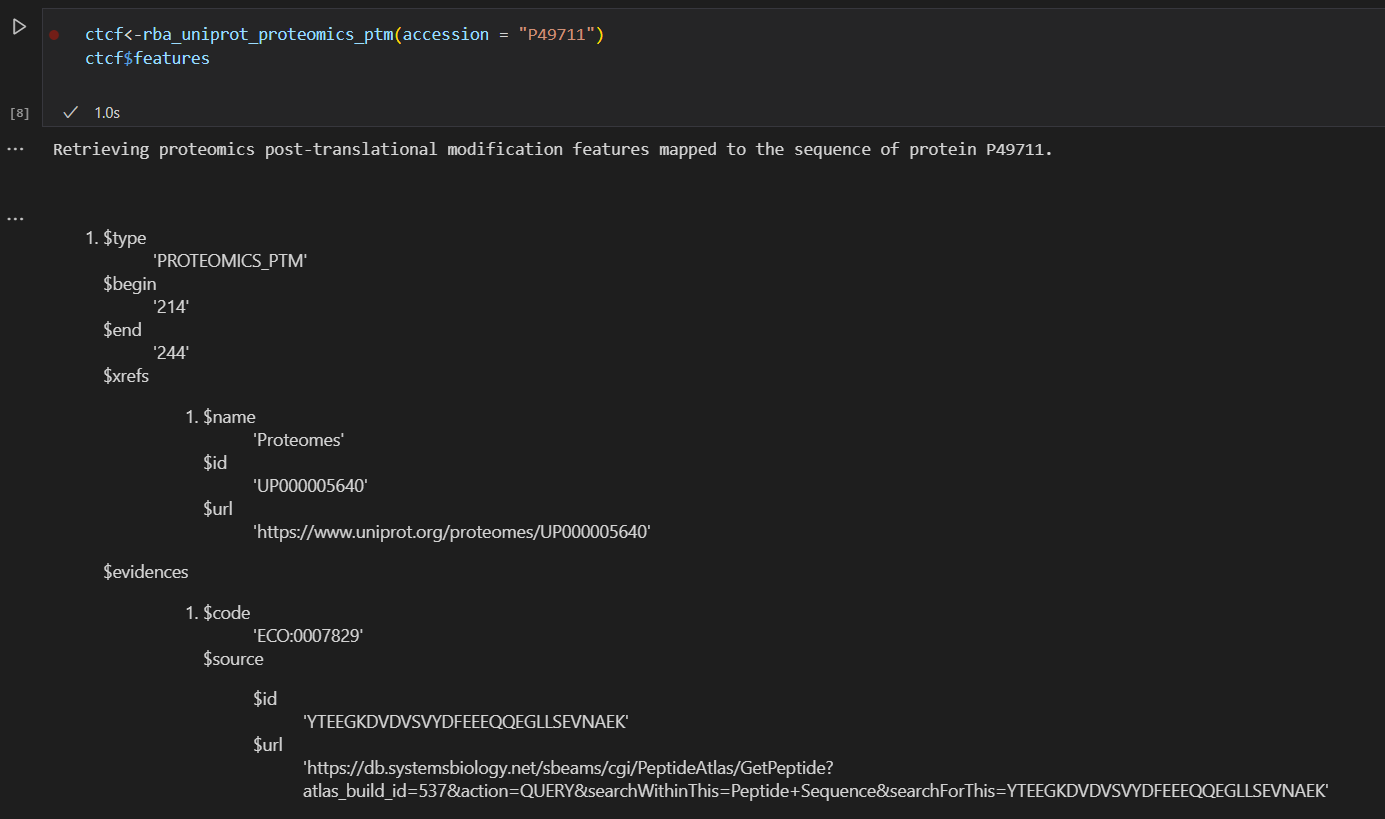
首先type、begin、end我们是要的,xref中的name和id我们也要,
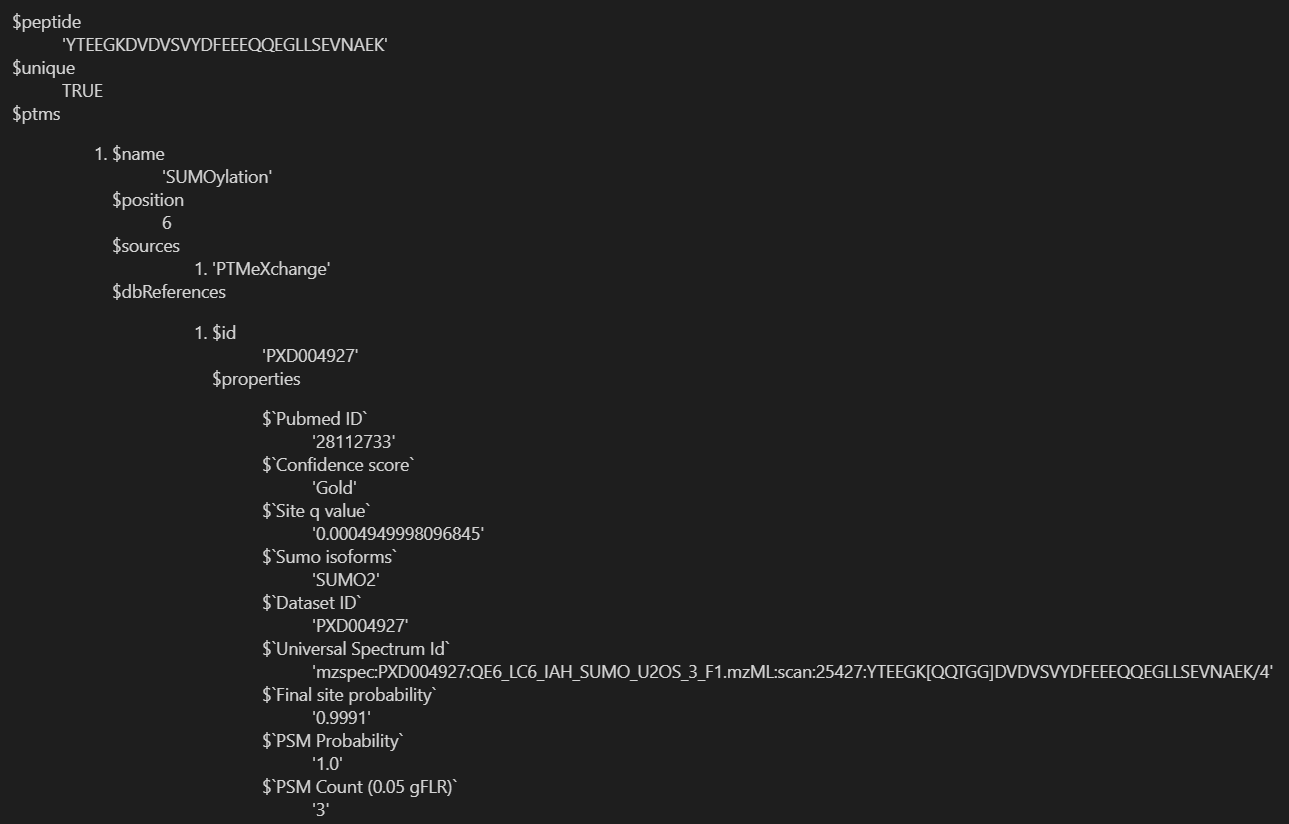
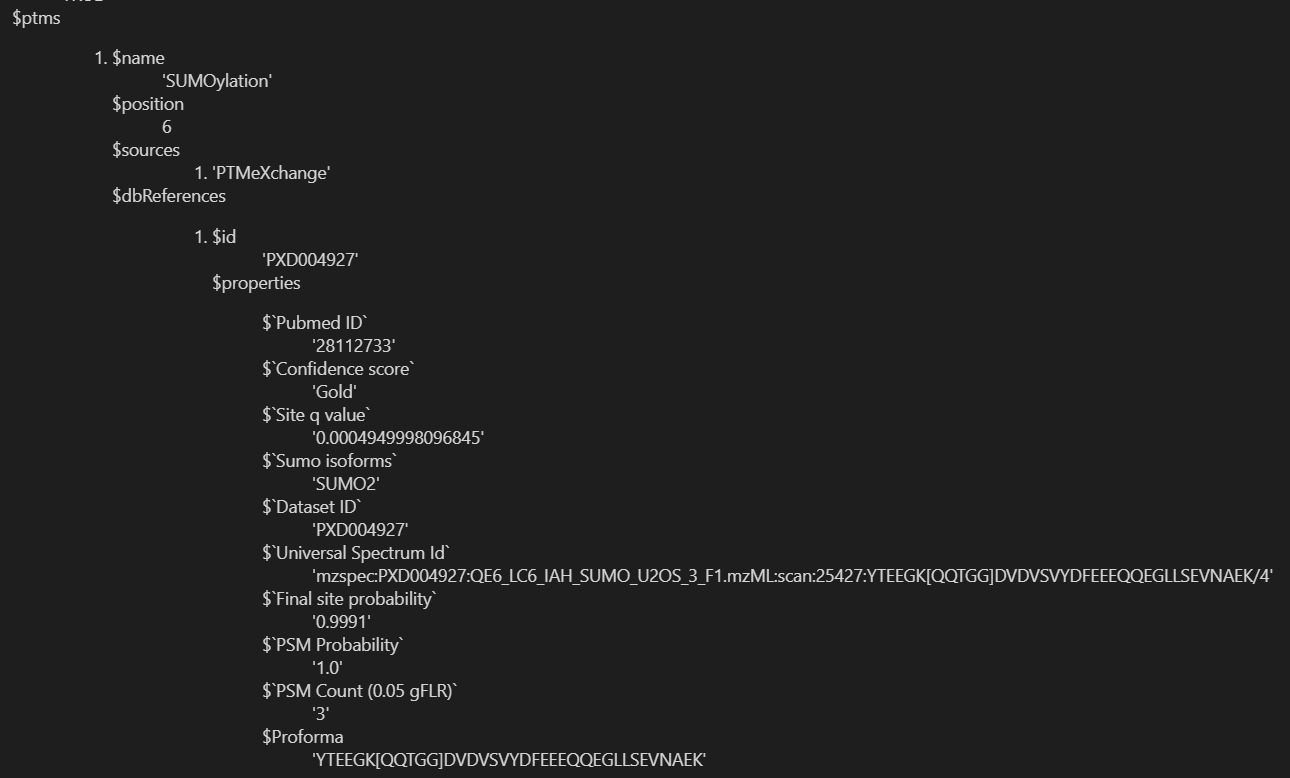
重点是下面这里的ptms:
我们要提取的是ptms的name、position、source,以及dbreference的id、properties的pubmed id,以及confidence score;
编写程序,获取其中的这些字段:
查看程序结果
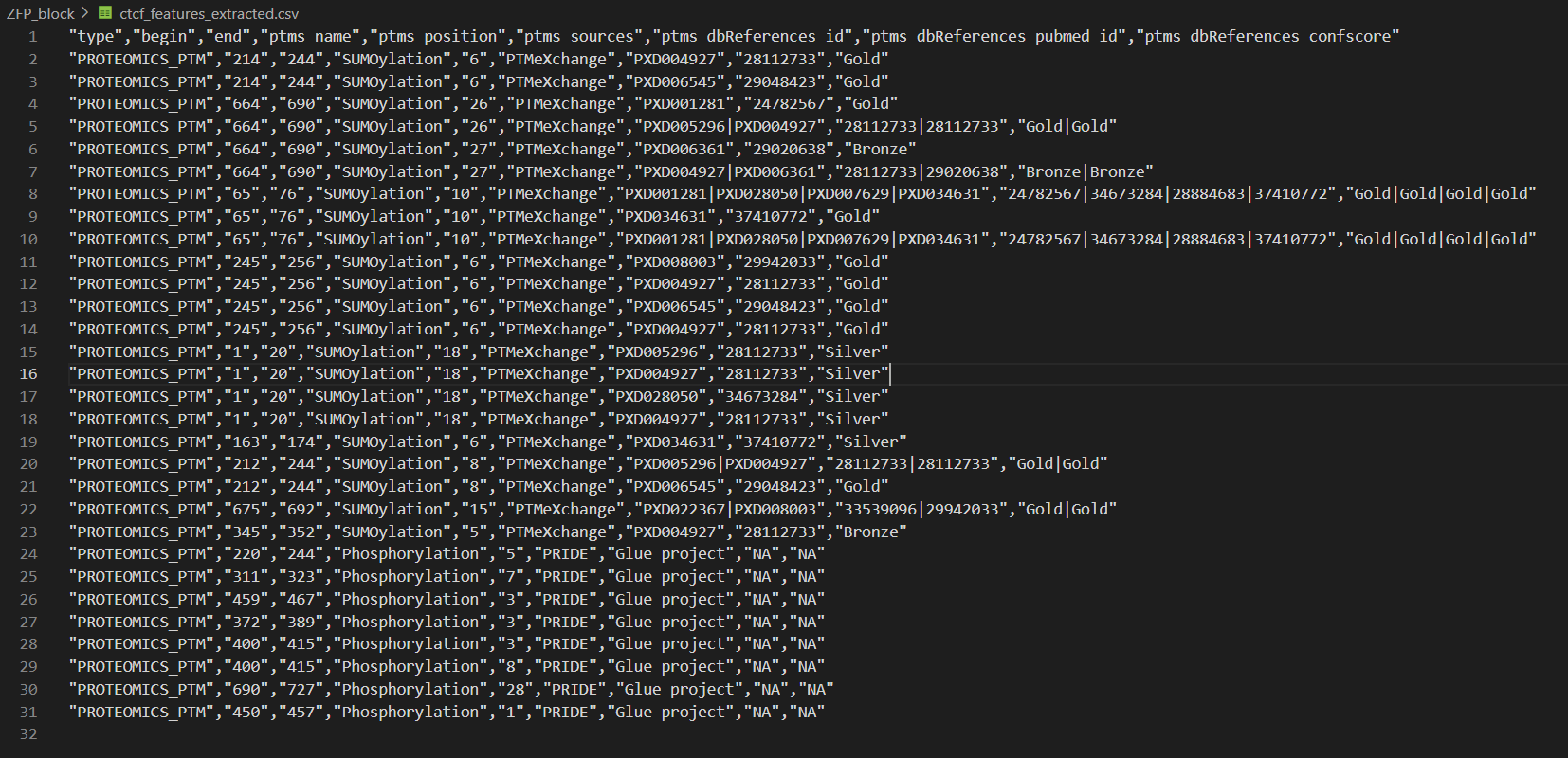
程序结果与获取的数据是没有问题的,
现在我们要返回去uniprot中查看这个数据结构对不对:
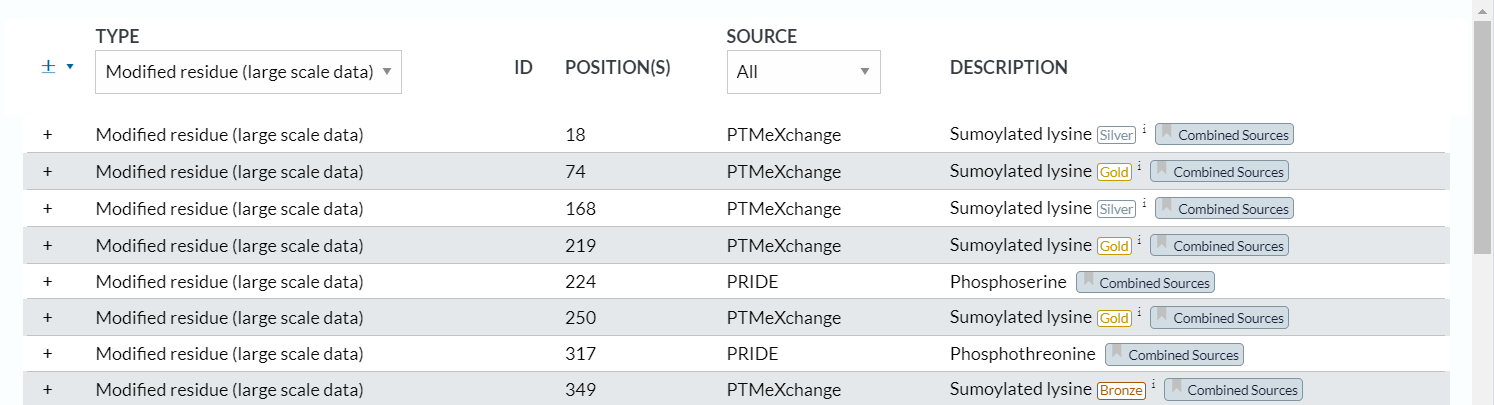
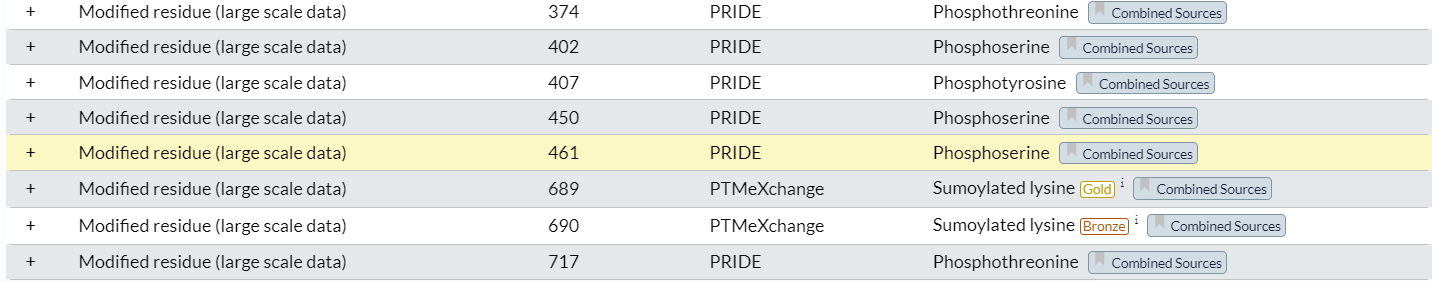
经过检查,确实这部分数据是完美匹配并且获取了large scale data中的PTM修饰
——》
那接下来的问题就是
①如何批量获取某个名单中的这些large scale data的PTM修饰数据
主要是前面单个的子函数要封装,然后后面就是遍历名单,写一个循环,然后批量使用上面的函数处理即可
我将这个代码的日志进行了记录: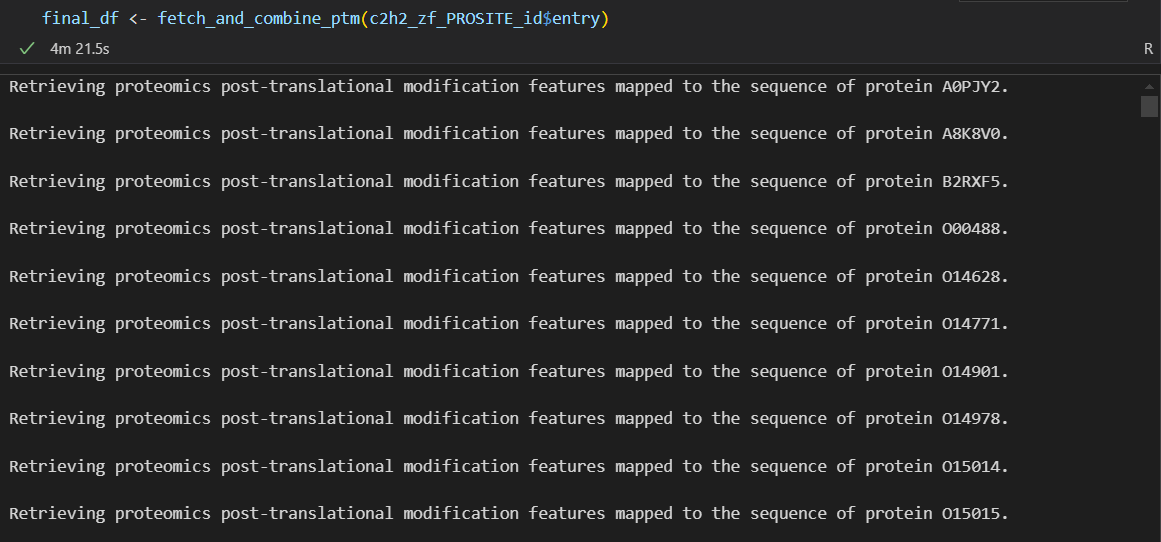
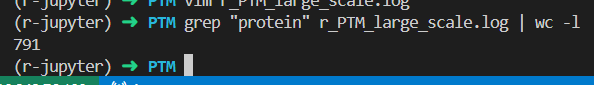
数目上还是一致的
统计了一下,暂时是
 749,也就是有42种蛋白质是没有大规模蛋白质组PTM数据的
749,也就是有42种蛋白质是没有大规模蛋白质组PTM数据的
我们需要用CTCF的数据来验证一下结果有没有问题:
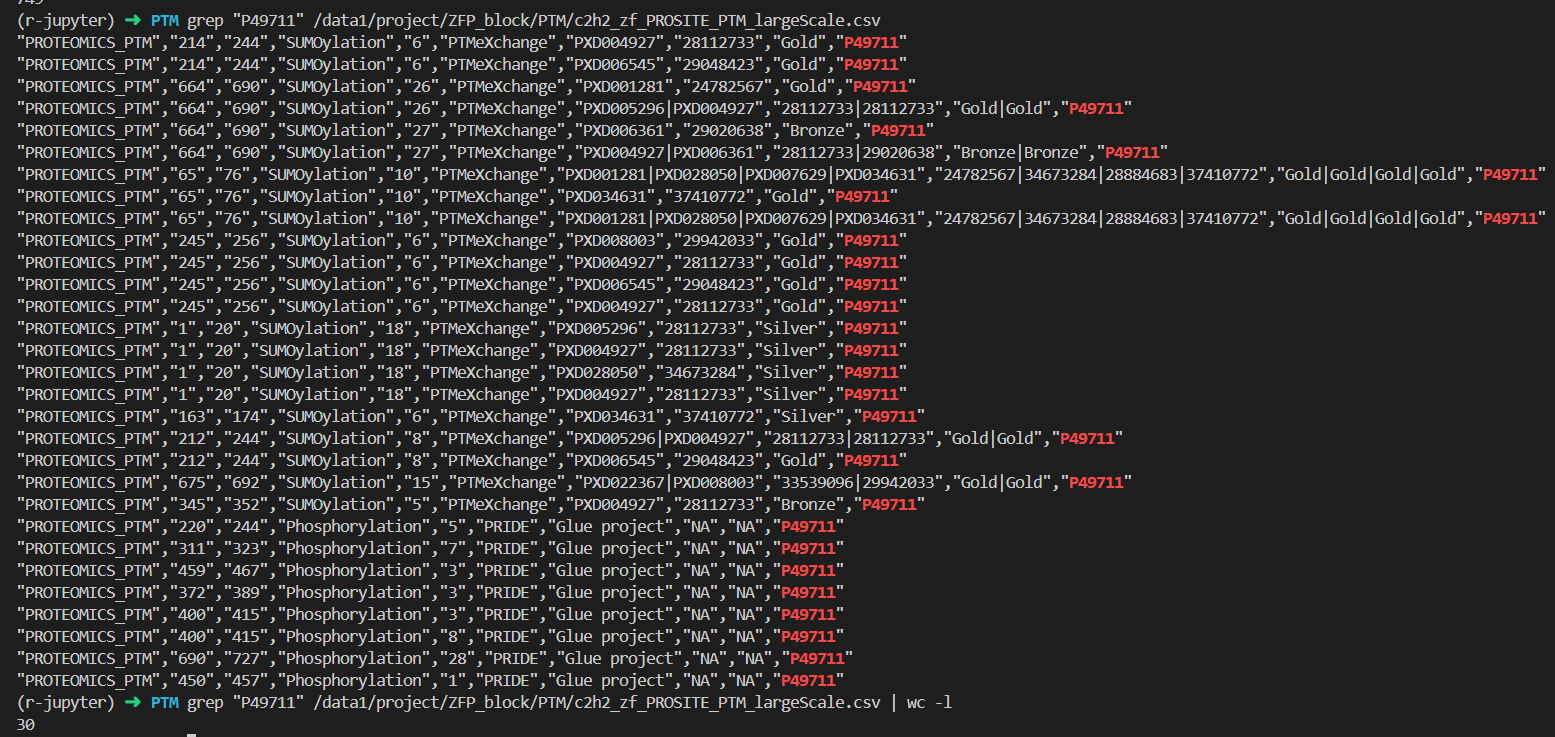
和前面的完全一致,所以是可以的
②如何将large scale data的数据与modi resi的文献uniprot的数据整合在一起
分开,前者用这个R包,后者uniprot数据自提;
都用这个包,因为数据是一起提取的;
四,额外数据库下载:
uniprot下载数据的时候还可以选择

实测没用,上面large scale data的还是下载不了


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









