






RNA-seq数据获取:Poly A plus RNA-seq (K562) GSM958729,获取BAM文件(GSM958729_hg19_wgEncodeCaltechRnaSeqK562R2x75Il200SplicesRep1V2.bam)。
PRO-seq数据获取:PRO-seq (K562) GSE166658,起初获取bigwig文件,尝试使用rtracklayer与genomicranges进行read counts获取,失败。后发现有作者做好的GSE166658_K562_PROseq_counts.csv文件,故直接调用。
BAM文件使用Subreads的featureCounts进行处理
新建虚拟环境
conda create -n env-name [list of package]-n env-name是设置新建环境的名字,list of package是可选项,选择要为该环境安装的包。
此处我创建的虚拟环境叫featurecounts。
conda activate featurecounts
conda install -c bioconda Subreads下载read counts需要对应的基因组gtf文件:GENCODE网站下载人类release 43 hg38文件。
参考:featurecounts的使用说明 - 简书 (jianshu.com)
参数说明
| 参数 | 说明 |
|---|---|
| input file | 输入的bam/sam文件,支持多个文件输入 |
| -a < string > | 参考gtf文件名,支持Gzipped文件格式 |
| -F | 参考文件的格式,一般为GTF/SAF,C语言版本默认的格式为GTF格式 |
| -A | 提供一个逗号分割为两列的文件,一列为gtf中的染色体名,另一列为read中对应的染色体名,用于将gtf和read中的名称进行统一匹配,注意该文件提交时不需要列名 |
| -J | 对可变剪切进行计数 |
| -G < string > | 当-J设置的时候,通过-G提供一个比对的时候使用的参考基因组文件,辅助寻找可变剪切 |
| -M | 如果设置-M,多重map的read将会被统计到 |
| -o < string > | 输出文件的名字,输出文件的内容为read 的统计数目 |
| -O | 允许多重比对,即当一个read比对到多个feature或多个metafeature的时候,这条read会被统计多次 |
| -T | 线程数目,1~32 |
| 下面是有关featrue/metafeature选择的参数 | 参数说明 |
| -p | 只能用在paired-end的情况中,会统计fragment而不统计read |
| -B | 在-p选择的条件下,只有两端read都比对上的fragment才会被统计 |
| -C | 如果-C被设置,那融合的fragment(比对到不同染色体上的fragment)就不会被计数,这个只有在-p被设置的条件下使用 |
| -d < int > | 最短的fragment,默认是50 |
| -D < int > | 最长的fragmen,默认是600 |
| -f | 如果-f被设置,那将会统计feature层面的数据,如exon-level,否则会统计meta-feature层面的数据,如gene-levels |
| -g < string > | 当参考的gtf提供的时候,我们需要提供一个id identifier 来将feature水平的统计汇总为meta-feature水平的统计,默认为gene_id,注意!选择gtf中提供的id identifier!!! |
| -t < string > | 设置feature-type,-t指定的必须是gtf中有的feature,同时read只有落到这些feature上才会被统计到,默认是“exon” |
featureCounts -a $gtf -o read.count -p -B -C -f -t exon -g gene_name *.bam对feature水平进行统计时,需要设置-f;对meta-feature水平进行统计时,不能设置-f。
使用中发现ucsc下载的gtf文件无法通过gene_name运行,故使用GENCODE下载的gtf文件。
随后对RNA-seq和PRO-seq的read counts进行TPM计算,可手算或使用r包脚本。
如果是用gene_name对BAM进行的read counts统计,则需要对gene symbol进行转换,至ensembl ID,从而获取每个基因对应的transpription unit长度。
使用biomaRt进行gene ID转换。
将gene symbol单拎出来建一个csv文件

在R studio中:
BiocManager::install("biomaRt")
library(biomaRt)
rg4 <- read.csv("gene symbol.csv",header = F,sep = ",")[,1]
mart = useMart(biomart = "ENSEMBL_MART_ENSEMBL",
dataset = "hsapiens_gene_ensembl",
host = "https://www.ensembl.org")
listFilters(mart)
s2esb = getBM(attributes = c("hgnc_symbol","ensembl_gene_id","chromosome_name","start_position","end_position"),filters = "hgnc_symbol",values = rg4,mart = mart)
write.table(as.data.frame(s2esb),"rg4-s2esb.tsv",sep = "\t",row.names = F,quote = F)
#至此为gene ID的转换,因为biomaRt要用ensembl的ID
feature_ids = read.csv("gene symbol-ensembl id.csv",header = T,sep = ",")[,2]
feature_info = biomaRt::getBM(attributes = c(
"hgnc_symbol",
"ensembl_gene_id",
"chromosome_name",
"start_position",
"end_position"),
filters = "ensembl_gene_id",values = feature_ids,mart = mart)
eff_length = abs(feature_info$end_position - feature_info$start_position)
names(eff_length) = feature_info$ensembl_gene_id
write.csv(eff_length,"rg4 gene_length.csv",row.names = TRUE)
#至此,输出gene lengthTPM计算:
手动用excel计算方法:

R脚本计算方法:
mycounts = read.csv("rna-seq&pro-seq reads 4 normalize.csv")
head(mycounts)
rownames(mycounts) = mycounts[,1]
mycounts<-mycounts[,-1]
head(mycounts)
kb <- mycounts$length / 1000
head(kb)
countdata <- mycounts[,1:2]
head(countdata)
rpk <- countdata / kb
head(rpk)
tpm = t(t(rpk)/colSums(rpk)*1000000)
head(tpm)
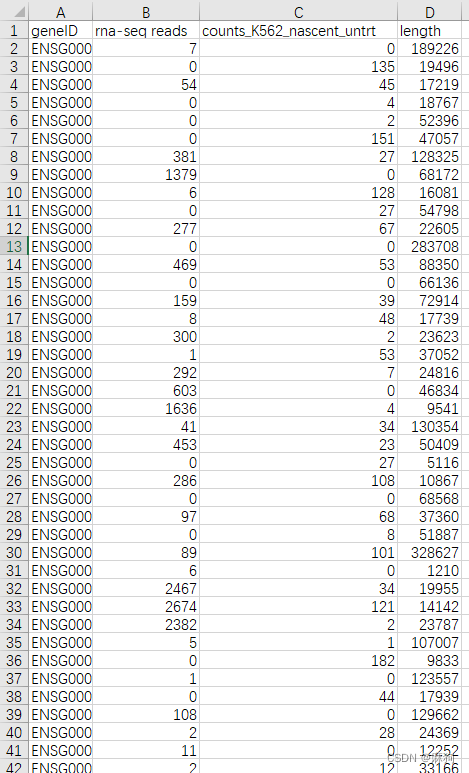
write.table(tpm,file = "rna-seq&pro-seq tpm.tsv",sep = "\t",quote = F)其中最先读入的csv文件整理成下面这样:















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









