






从键盘输入一个纯净物的化学式,输出其分子量,结果保留3位小数。输入占1行;输出占1行。

def sum(element_counts, elements_dict):
total_weight = 0
for element, count in element_counts.items():
total_weight += elements_dict[element] * count
return total_weight
def get_molecular_weight(formula):
# 定义element_counts作为字典来存储元素及其数量
element_counts = {}
# 定义一个栈用于存储开括号的位置
stack = []
# 定义元素符号和对应的原子量
elements_dict = {
'H': 1.007,
'He': 4.002,
'Li': 6.938,
'Be': 9.012,
'B': 10.806,
'C': 12.0096,
'N': 14.006,
'O': 15.999,
'F': 18.998,
'Ne': 20.1797,
'Na': 22.989,
'Mg': 24.305,
'Al': 26.981,
'Si': 28.084,
'P': 30.973,
'S': 32.059,
'Cl': 35.45,
'K': 39.0983,
'Ca': 40.078,
'Sc': 45.0,
'Ti': 47.9,
'V': 50.9,
'Cr': 51.9961,
'Mn': 54.938,
'Fe': 55.845,
'Co': 58.9,
'Ni': 58.7,
'Cu': 63.6,
'Zn': 65,
}
i = 0
while i < len(formula):
if formula[i].isupper():
element = formula[i]
i += 1
while i < len(formula) and formula[i].islower():
element += formula[i]
i += 1
count = ''
while i < len(formula) and (formula[i].isdigit() or formula[i] == '/'):
count += formula[i]
i += 1
if count == '':
count = '1'
element_counts[element] = int(count)
elif formula[i] == '(':
stack.append(i)
i += 1
elif formula[i] == ')':
start = stack.pop()
end = i
num = ''
i += 1
while i < len(formula) and (formula[i].isdigit() or formula[i] == '/'):
num += formula[i]
i += 1
if num == '':
num = '1'
sub_formula = formula[start + 1:end]
sub_weight = get_molecular_weight(sub_formula)
for j in range(start + 1, end):
if formula[j].isupper():
element_counts[formula[j]] *= int(num)
for element, count in sub_weight.items():
element_counts[element] = element_counts.get(element, 0) + count
else:
i += 1
return element_counts
# 获取输入的化学式
m = input()
# 去除空格
list0 = ''.join(m.split())
# 定义元素的原子量字典
elements_dict = {
'H': 1.007,
'He': 4.002,
'Li': 6.938,
'Be': 9.012,
'B': 10.806,
'C': 12.0096,
'N': 14.006,
'O': 15.999,
'F': 18.998,
'Ne': 20.1797,
'Na': 22.989,
'Mg': 24.305,
'Al': 26.981,
'Si': 28.084,
'P': 30.973,
'S': 32.059,
'Cl': 35.45,
'K': 39.0983,
'Ca': 40.078,
'Sc': 45.0,
'Ti': 47.9,
'V': 50.9,
'Cr': 51.9961,
'Mn': 54.938,
'Fe': 55.845,
'Co': 58.9,
'Ni': 58.7,
'Cu': 63.6,
'Zn': 65,
}
# 调用get_molecular_weight函数计算分子量
molecular_weight = sum(get_molecular_weight(list0), elements_dict)
print('%.3f' % molecular_weight)测试运行结果:
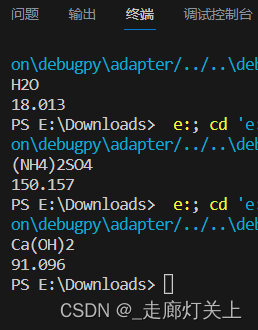
注意,某些复杂化学式如络合物等的分子式无法计算。



















 976
976

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











