







GaussianView5.0+Gaussian09,不得不说,这两个软件简直就是量化计算的神器!但是网上提供的教程并不多,一些专业知识我也是刚刚从论文中获得的。我现在用到了就是通过GaussianView5.0绘制分子结构,调整分子结构,编写脚本程序,扔到Gaussian软件中算。
Step1:画分子初始结构
在GaussianView软件中绘制水分子结构,百度有一些教程,首先选择成键的类型、然后选择相应元素的原子往键上放。为了方便后面调整结构,可以在GaussianView中点击View→Labels,给每个原子编号。
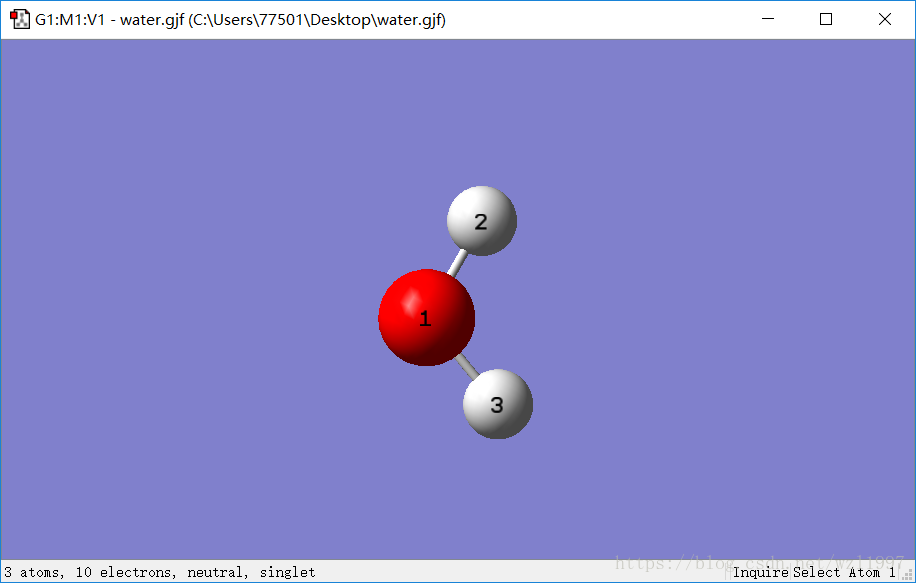
Step2:生成内坐标
在GaussianView把原子信息表调出来,查看笛卡尔坐标、Z-矩阵......

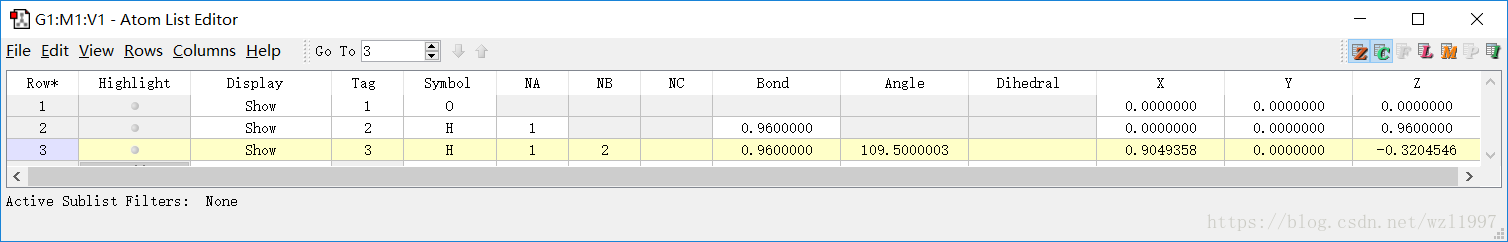
通常在计算原子比较多的较大分子时,如果需要改变分子结构,还需要消除原子之间的依赖性:可以通过手工修改原子编号。造成这种冲突的原因是因为某些分子的对称性。
然后添加扫描想信息:打开Scan界面:
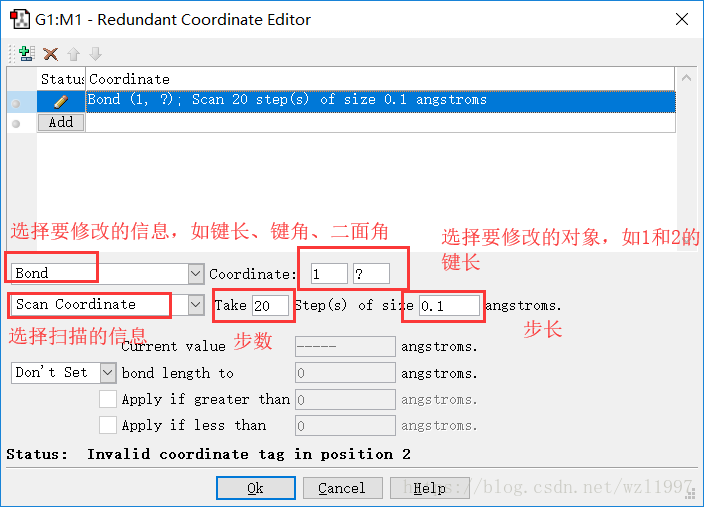
点击OK后,把鼠标移至紫色框的“G1:M1:V1”中,用Ctrl+S保存,主要保存时,要取消Write Cartesians的勾选。
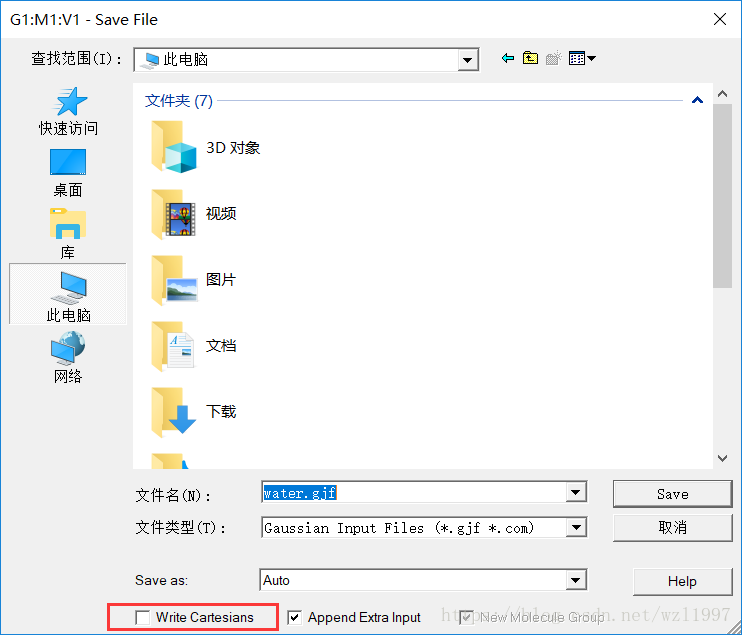
此时,H20的内坐标文件已经生成
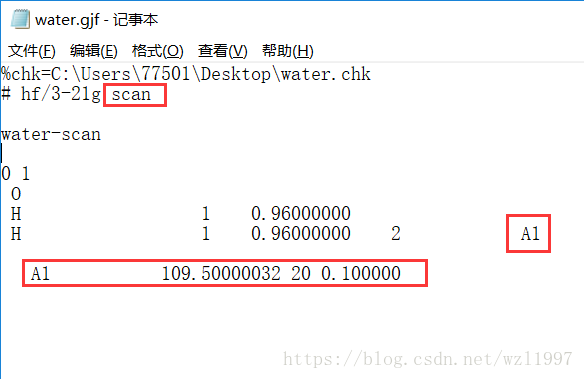
但是要注意的时,这个内坐标文件有时候并不是最后可以运行的文件,对于我此次试验来说就不是的。。。首先,要改一下计算单点能的方法,通常有HF、DFT、MP2、MP4等量化计算方法,这些都是围绕量子力学而发展的,求解薛定谔方程(你懂得。。)这里我选择了HF,因为需要的CPU和内存不多(在实验室的话一般就在服务器上跑了)。后面的3-21g是基组,基组有很多,我看的论文有很多都是基于CCSD(T)的,这个需要的计算量也比较大。
Step3:Scan一下
在Gaussian09中选择file→open打开生成的.gjf内坐标文件。打开后可以按下右上的运行按钮,如果有错误的话,软件会在第一时间报错。
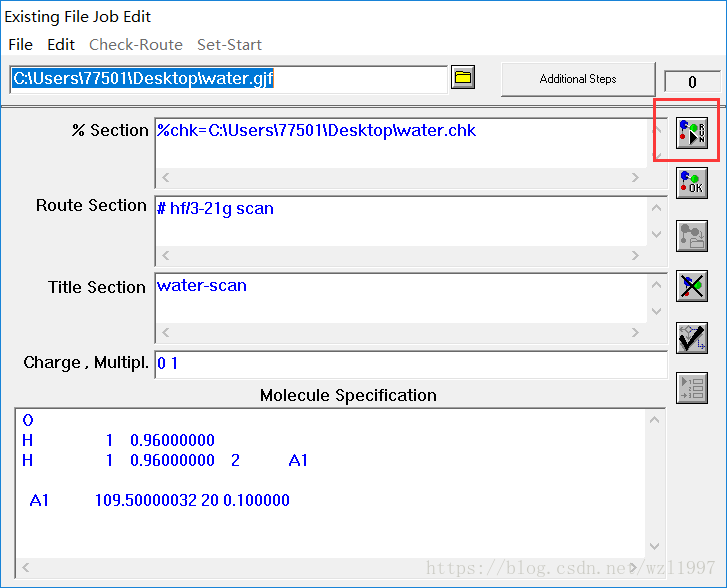
结束的标志就是“Normal”

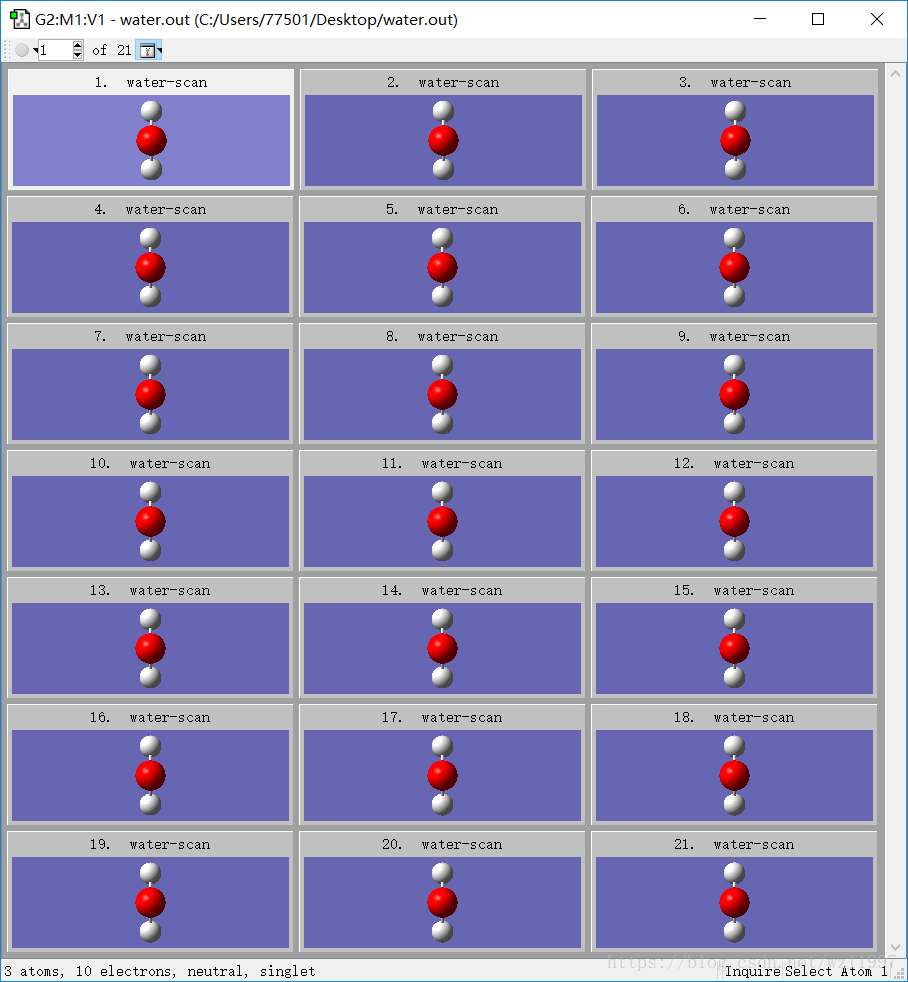
由于H2O分子体系相对小,并且我只修改了其中一个键长,步数只有20,所以运行很快,1分钟就OK了。
Step4:扫描结果
不算初始的参数,步数为20,总共有21种,构型如下(看起来变化不大,步长太小):
点击Results→Scan..,可以看到分子势能随键长的变化图,右键该图,save data到本地中,利用Origin软件作出该带点折线图。
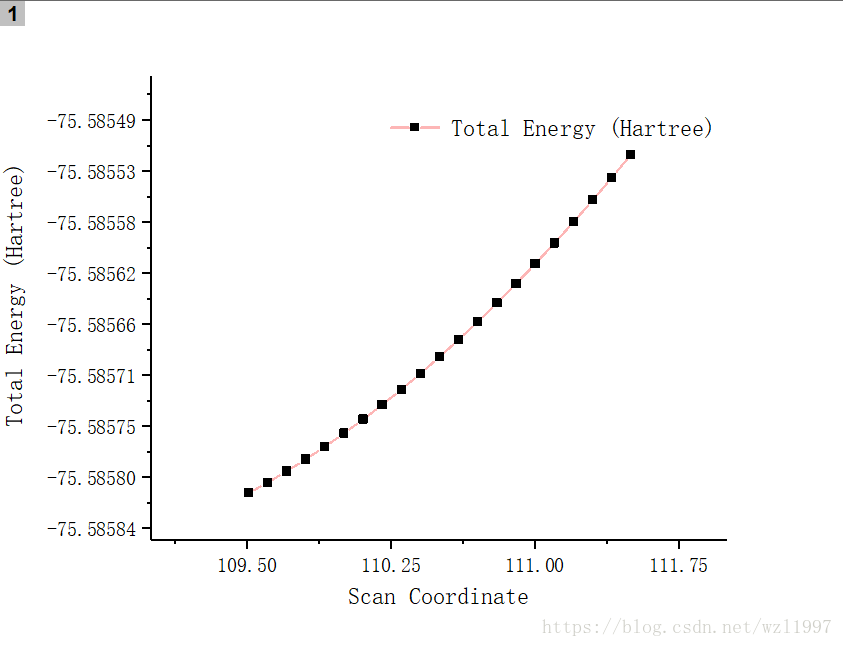
从该图可以看出,随着该键长的增大,势能在增加, 但并不能看出极小值点和极大值点。因此还需要修改分子构型,改变另一个键长和分子的键角,获得更多的单点能。可想而知,对于大分子的体系,自由度大之大,其计算量之大。


















 1717
1717

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











