







2021SC@SDUSC
文章目录
前言
AlphaFold2的代码真的是太多了,而且几乎没有注释,难度真的很大。因为我对深度学习的了解也并不深刻,并且TensorFlow这个框架也没有用过,所以在看代码的过程,真的是不停地在学习。总之一句话就是痛并快乐着。
今天要学习的是alphafold/alphafold/model/modules.py。modules.py 中主要是AlphaFold2 核心部分使用的模块和代码。
一、各种import
1、代码
import functools
from alphafold.common import residue_constants
from alphafold.model import all_atom
from alphafold.model import common_modules
from alphafold.model import folding
from alphafold.model import layer_stack
from alphafold.model import lddt
from alphafold.model import mapping
from alphafold.model import prng
from alphafold.model import quat_affine
from alphafold.model import utils
import haiku as hk
import jax
import jax.numpy as jnp
2、解析
1、导入了很多alphafold自己的模块
下面是各个模块的用途:
residue_constants.py中写明了AlphaFold 中使用的常量
all_atom.py中写明了所有原子表示的操作
common_modules.py中写明了用于蛋白质折叠的常见俳句模块的集合
folding.py中写明了结构模块的模块和实用程序
layer_stack.py中写明了没有共享参数的堆叠层函数重复的函数
lddt.py中写明了lDDT 蛋白质距离评分
mapping.py中写明了专门的映射函数
prng.py中写明了关于蛋白质折叠中 PRNG 使用的实用程序集合
quat_affine.py中写明了四元数几何模块
utils.py中写明了一组用于蛋白质折叠的 JAX 实用函数
2、导入了很多库
Haiku:
Haiku是JAX的神经网络库,它允许用户使用熟悉的面向对象编程模型,同时允许完全访问JAX的纯函数转换。它提供了两个核心工具:模块抽象hk.Module,和一个简单的函数转换hk.transform。
hk.Module是Python对象,包含对其自身参数、其他模块和对用户输入应用函数方法的引用。hk.transform允许完全访问JAX的纯函数转换。在后面的代码中就用到了hk.Module。
functools:
functools是python的模块,用于高阶函数:指那些作用于函数或者返回其它函数的函数,通常只要是可以被当做函数调用的对象就是这个模块的目标。
cmp_to_key,将一个比较函数转换关键字函数;
partial,针对函数起作用,并且是部分的;
reduce,与python内置的reduce函数功能一样;
total_ordering,在类装饰器中按照缺失顺序,填充方法;
update_wrapper,更新一个包裹(wrapper)函数,使其看起来更像被包裹(wrapped)的函数;
wraps,可用作一个装饰器,简化调用update_wrapper的过程;
二、softmax_cross_entropy函数
1、代码
def softmax_cross_entropy(logits, labels):
loss = -jnp.sum(labels * jax.nn.log_softmax(logits), axis=-1)
return jnp.asarray(loss)
2、解析
上述代码是在给定 logits 和 one-hot class 标签的情况下计算 softmax 交叉熵
softmax函数,又称归一化指数函数。它是二分类函数sigmoid在多分类上的推广,目的是将多分类的结果以概率的形式展现出来。
交叉熵(Cross Entropy)是Shannon信息论中一个重要概念,主要用于度量两个概率分布间的差异性信息。
有关softmax函数的详解,请见https://zhuanlan.zhihu.com/p/105722023
有关交叉熵的详解,请见https://blog.csdn.net/qq_33542428/article/details/106482393
三、sigmoid_cross_entropy函数
1、代码
def sigmoid_cross_entropy(logits, labels):
log_p = jax.nn.log_sigmoid(logits)
# log(1 - sigmoid(x)) = log_sigmoid(-x), the latter is more numerically stable
log_not_p = jax.nn.log_sigmoid(-logits)
loss = -labels * log_p - (1. - labels) * log_not_p
return jnp.asarray(loss)
2、解析
上述代码在给定 logits 和多个类标签的情况下计算 sigmoid 交叉熵
sigmoid函数表达式和图像如下:
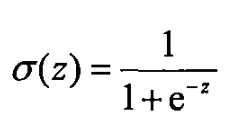
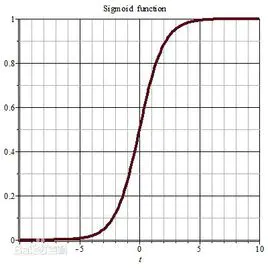
可以看到在趋于正无穷或负无穷时,函数趋近平滑状态,sigmoid函数因为输出范围(0,1),所以二分类的概率常常用这个函数,这个函数有以下几个优点:
(1)值域在0和1之间
(2)函数具有非常好的对称性
(3)函数对输入超过一定范围就会不敏感
有关sigmoid函数的详解,请见https://www.jianshu.com/p/506595ec4b58
四、create_extra_msa_feature函数
1、代码
def create_extra_msa_feature(batch):
# 23 = 20 amino acids + 'X' for unknown + gap + bert mask
msa_1hot = jax.nn.one_hot(batch['extra_msa'], 23)
msa_feat = [msa_1hot,
jnp.expand_dims(batch['extra_has_deletion'], axis=-1),
jnp.expand_dims(batch['extra_deletion_value'], axis=-1)]
return jnp.concatenate(msa_feat, axis=-1)
2、解析
将 extra_msa 扩展为 one_hot并与其他额外的 msa 功能连接。
尽可能晚地执行此操作,因为 one_hot 额外的 msa 可能非常大。
参数:批处理:具有以下键的字典:
extra_msa: [N_extra_seq, N_res] 未被选为集群的 MSA中心。这不是one_hot编码。
extra_has_deletion: [N_extra_seq, N_res] 附加MSA的每个位置的左侧是否有删除。
extra_deletion_value: [N_extra_seq, N_res] 附加MSA的每个位置左侧的删除次数。
返回:附加MSA特征的串联张量。
五、AlphaFoldIteration类
1.1、代码
class AlphaFoldIteration(hk.Module):
def __init__(self, config, global_config, name='alphafold_iteration'):
super().__init__(name=name)
self.config = config
self.global_config = global_config
1.2、解析
AlphaFoldIteration类:
AlphaFoldIteration类的主要作用是实现了AlphaFold 架构的单次循环迭代
根据提供的特征计算集成(平均)表示。
然后将这些表示传递给配置文件请求的各个头。
每个头还返回一个损失,该损失作为加权和组合,以产生总损失。
2.1、代码
evoformer_module = EmbeddingsAndEvoformer(
self.config.embeddings_and_evoformer, self.global_config)
batch0 = slice_batch(0)
representations = evoformer_module(batch0, is_training)
2.2、解析
计算每个批次元素和平均值的表示
3.1、代码
msa_representation = representations['msa']
del representations['msa']
3.2、解析
MSA 表示没有集成,所以我们不将张量传递到循环中。
4.1、代码
if ensemble_representations:
def body(x):
i, current_representations = x
feats = slice_batch(i)
representations_update = evoformer_module(
feats, is_training)
new_representations = {}
for k in current_representations:
new_representations[k] = (
current_representations[k] + representations_update[k])
return i+1, new_representations
if hk.running_init():
_, representations = body((1, representations))
else:
_, representations = hk.while_loop(
lambda x: x[0] < num_ensemble,
body,
(1, representations))
for k in representations:
if k != 'msa':
representations[k] /= num_ensemble.astype(representations[k].dtype)
4.2、解析
上述代码是对批处理维度上的表示(MSA 除外)求平均。
其中def body(x)后面是将一个元素添加到表示集合中去。
5.1、代码
if hk.running_init():
_, representations = body((1, representations))
else:
_, representations = hk.while_loop(
lambda x: x[0] < num_ensemble,
body,
(1, representations))
5.2、解析
上述代码是初始化Haiku 模块时,运行while_循环的一次迭代来初始化“body”中使用的Haiku 模块。
6.1、代码
for name, (head_config, module) in heads.items():
if name in ('predicted_lddt', 'predicted_aligned_error'):
continue
else:
ret[name] = module(representations, batch, is_training)
if 'representations' in ret[name]:
representations.update(ret[name].pop('representations'))
if compute_loss:
total_loss += loss(module, head_config, ret, name)
6.2、解析
跳过 PredictedLDDTHead 和 PredictedAlignedErrorHead 直到StructureModule 被执行。
结构模块用来自头部的额外表示为PredictedLDDTHead提供激活。
7.1、代码
if self.config.heads.get('predicted_lddt.weight', 0.0):
name = 'predicted_lddt'
head_config, module = heads[name]
ret[name] = module(representations, batch, is_training)
if compute_loss:
total_loss += loss(module, head_config, ret, name, filter_ret=False)
7.2、解析
在 StructureModule 执行后添加 PredictedLDDTHead
提供所有以前的结果以访问 structure_module 结果。
8.1、代码
if ('predicted_aligned_error' in self.config.heads
and self.config.heads.get('predicted_aligned_error.weight', 0.0)):
name = 'predicted_aligned_error'
head_config, module = heads[name]
ret[name] = module(representations, batch, is_training)
if compute_loss:
total_loss += loss(module, head_config, ret, name, filter_ret=False)
8.2、解析
在 StructureModule 执行后添加 PredictedAlignedErrorHead。
提供所有以前的结果以访问 structure_module 结果。














 1019
1019











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









