






 FINEMAP是一种利用GWAS汇总统计数据进行精细映射的程序,旨在识别和估计因果SNPs及其效应大小和遗传力分布。它采用shotgunstochasticsearch算法,对大规模数据有高计算效率和准确性。FINEMAP输出包括潜在因果配置的后验概率和贝叶斯因子,以及SNP的因果性概率,是基因组研究的重要工具。
FINEMAP是一种利用GWAS汇总统计数据进行精细映射的程序,旨在识别和估计因果SNPs及其效应大小和遗传力分布。它采用shotgunstochasticsearch算法,对大规模数据有高计算效率和准确性。FINEMAP输出包括潜在因果配置的后验概率和贝叶斯因子,以及SNP的因果性概率,是基因组研究的重要工具。
这篇笔记主要是介绍FINEMAP,参见网页。
有关FINEMAP的文章:
Refining fine-mapping: effect sizes and regional heritability. bioRxiv. (2018).
Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Hum. Genet. (2017).
FINEMAP: Efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493-1501 (2016).
芬兰赫尔辛基大学 GWAS 课程:https://www.mv.helsinki.fi/home/mjxpirin/GWAS_course/
FINEMAP是什么
是一个能够识别causal SNPs,能够估计causal SNPs效应大小,以及估计causal SNPs遗传力分布的程序。主要有MacOS X 和Unix两个版本,可以从上述网址中下载。
在与复杂性状和疾病相关的基因组区域,FINEMAP通过使用来自全基因组关联研究的汇总统计数据,具有很高的计算效率,而且FINEMAP通过应用shotgun stochastic search(SSS)霰弹枪随机搜索算法,具有鲁棒性(Hans等人,2007)。与现有方法相比,产生的结果较为精确,耗时很少。因此,是对全基因组关联研究和新兴测序或生物库项目中产生的大量数据进行分析的理想工具。FINEMAP 的优点是,在设定好最大causal SNPs 数量 k 后,得到的结果包含了 1 到 k 个不同 causal SNPs 的后验概率以及各个 causal SNPs 数量的概率,非常方便进行后续的分析。
fine-mapping的基本流程
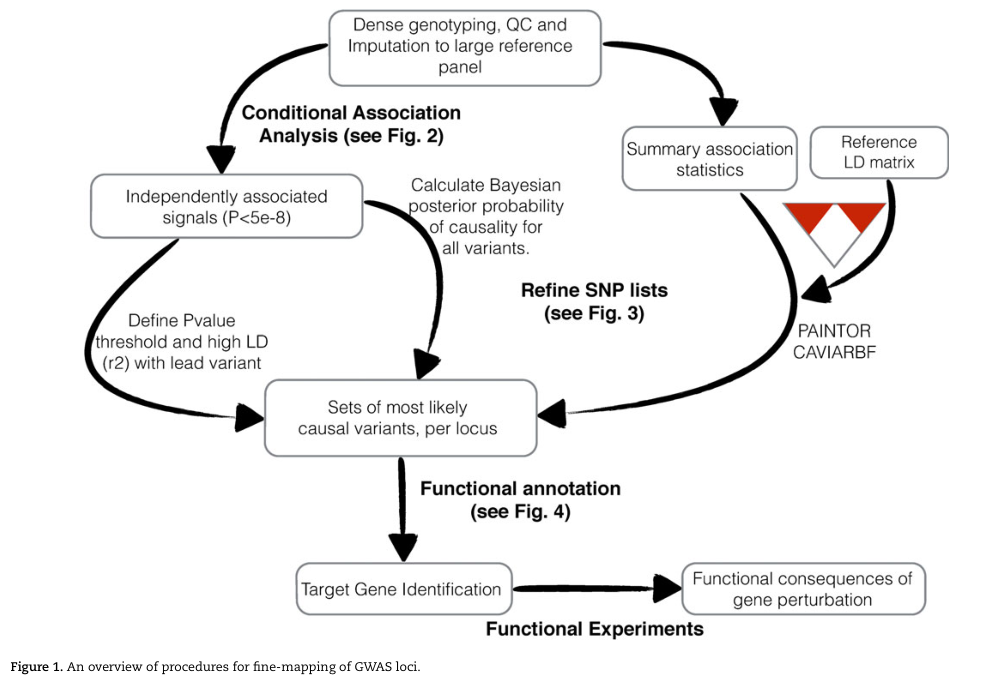
Spain SL et al(2015),Hum Mol Genet.
fine-mapping的LD可以来源于原始的表型和基因型数据,也可以由reference panel提供,但是用reference panel提供的LD需要多大的样本量去实现相当于原始GWAS 数据产生的LD accuracy?值得考证
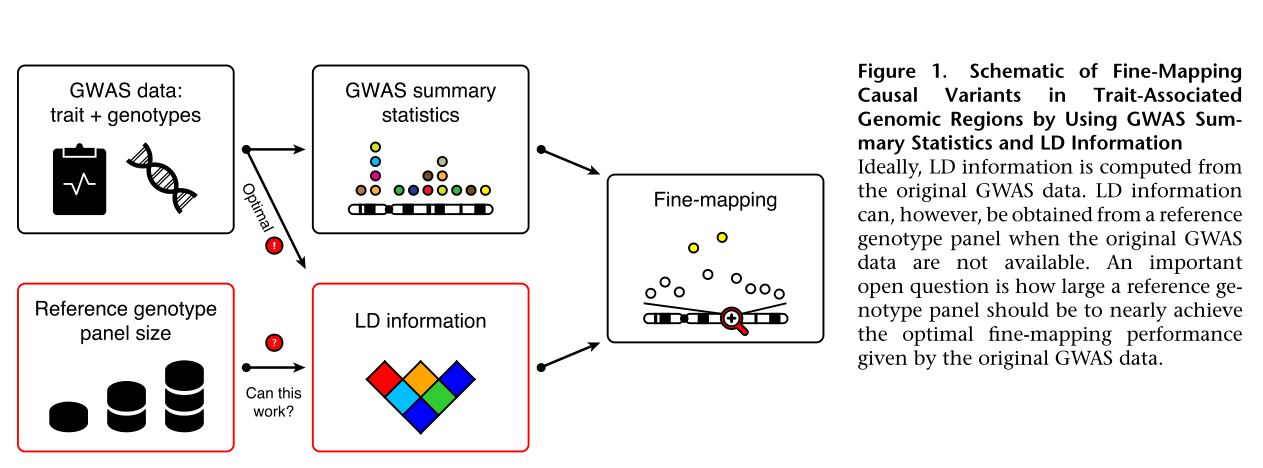
In this paper researchers established that for a typical GWAS cohort containing up to 10,000 individuals, a reference panel of 1,000 individuals from the study population (Finland or the UK in our examples) is adequate, whereas a reference panel of about 100 individuals from the study population (e.g.,1000GP data) is too small and should not be used.
用下图来说明LD和LD block
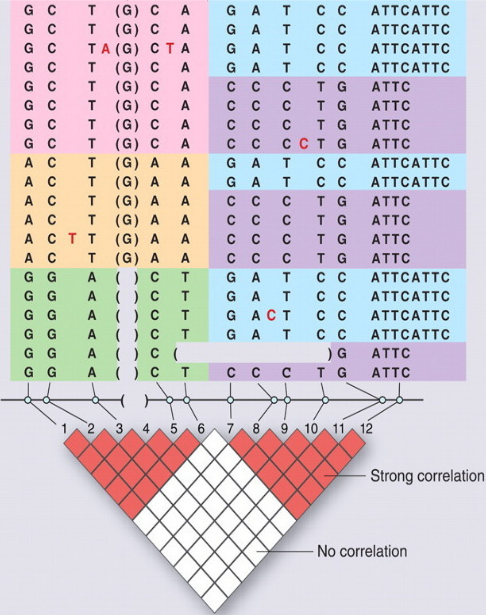
Altshuler et al. (2008)
目前fine-mapping的方法
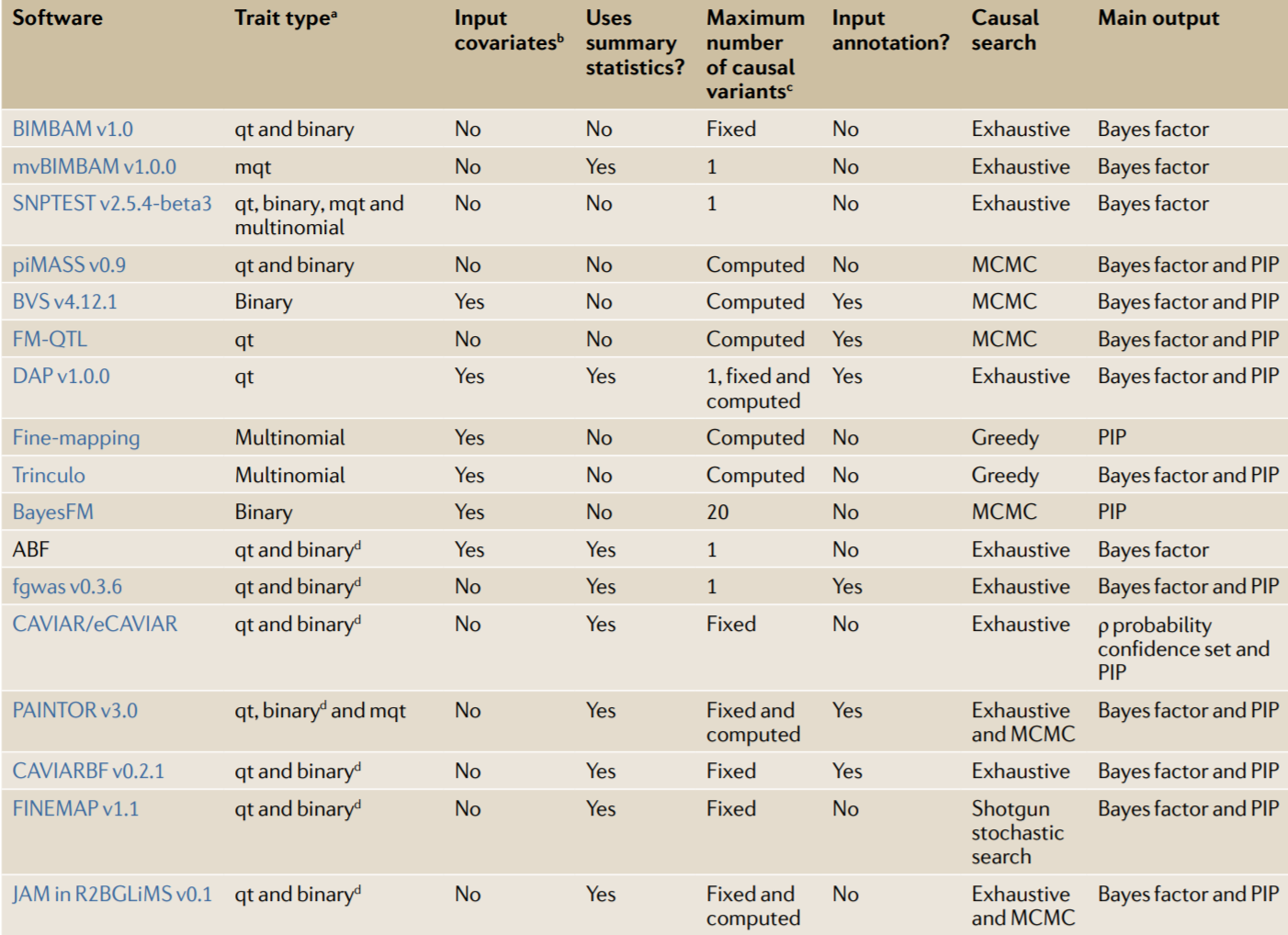
Schaid et al. (2018) Nat. Rev. Genet.
命令行参数说明
--cond 通过逐步条件回归来进行fine-mapping
--cond-pvalue 设置全基因组显著性水平的p值,默认是5 × 10-8
--corr-config 如果因果configuration包含一对绝对相关性高于此阈值的 SNP,则可选择将因果配置的后验概率设置为零,默认是0.95。
--dataset 用于指定以分隔符分隔的数据集列表的选项,以便在master文件中进行fine-mapping(例如 1、2 或 1|2表示第一行或第二行),默认情况下处理所有数据集。
--flip-beta 用于读取 Z 文件中的列“flip”的选项,其中包含二进制指示符,指定是否需要翻转估计的 SNP 效应大小的方向以匹配 SNP 相关性,结合 --cond, --config 和--sss。
--force-n--samples 允许在一组大小与 GWAS 样本数量不同的样本上计算 BCOR 文件中的相关性的选项,结合 --cond, --config 和--sss。
--help 命令行帮助。
--in-files master 文件。结合 --cond, --config 和--sss。
--log 用于将输出写入master文件“log”列中指定的日志文件的选项,默认情况下不写入任何日志文件。
--n-causal-snps 设置允许最多causal SNPs的数目,默认是5。
--n-configs-top 设置要保存的top因果configuration的数量的选项,默认是50000。
--n-conv-sss 用于设置在霰弹枪随机搜索终止之前,添加的概率mass需要低于指定阈值 (--prob-conv-sss-tol) 的迭代次数的选项,默认是100。
--n-iter 用于设置在霰弹枪随机搜索终止之前的最大迭代次数的选项,默认是100000。
--n-threads 设置并行线程数的选项,默认是1。
--prior-k 选择对master文件中 K 文件(见下文)中指定的因果 SNP 数使用先验概率,默认情况下,SNP被假定为因果关系,概率为1 /(基因组区域中的SNP数量)。
--prior-k0 设置基因组区域中没有因果 SNP 的先验概率的选项。仅在计算因果SNP数量的后验概率时使用,但在fine-mapping期间不用。默认是0.0。
--prior-snps 用于读取 Z 文件中具有 SNP 是因果关系的先验概率的“概率”列的选项,以便为每个因果configuration定义先验概率,结合--sss。
--prior-std 用于指定以逗号分隔的效应大小的先验标准差列表的选项,默认是0.05。
--prob-conv-sss-tol 用于设置容差的选项,在容差下,添加的概率质量(在 --n-conv-sss 迭代上)被认为足够小,以终止霰弹枪随机搜索。默认是0.001。
--prob-cred-set 设置可信区间包含因果 SNP 的概率的选项,默认是0.95。
--pvalue-snps 用于设置包含 SNP 的 p 值阈值的选项,默认是1。
--rsids 用于分离与 Z 文件中的 rsid 列相对应的逗号分隔的 SNP 标识符列表的选项(见下文),结合--config。
--sss 使用霰弹枪随机搜索进行fine-mapping。
--std-effects 可选择输出标准化dosage后效应大小分布的均值和标准差,默认是allele dosage。
输入说明
master文件
主文件是以分号分隔的文本文件,不包含空格。它包含以下必需列名和每行一个数据集。
z 列包含 Z 文件的名称(输入)
ld 列包含 LD 文件的名称(输入)
bcor 列包含 BCOR 文件的名称(输入)
snp 列包含 SNP 文件的名称(输出)
config列包含配置文件(输出)的名称
cred列包含 CRED 文件的名称(输出)
n_samples列包含 GWAS 样本数量
k 列包含可选 K 文件的名称(可选输入)
log列包含可选 LOG 文件的名称(可选输出)
文件扩展名必须与标题行中的列名相对应!
master文件可以同时包含 ld 和 bcor 列。对于每行的每个数据集,需要为预先计算的 SNP 相关性指定条目,这些相关性以文本格式显示在列 ld 或列 bcor 中以二进制 fomat 表示。如果一行在两列中都包含条目,则使用预先计算的 SNP 相关性。
如果在一组大小与列n_samples中指定的样本不同,则 --force-n-samples 命令行参数可用于覆盖错误检查。
具有两个使用预先计算的 SNP 相关性的数据集的master文件应如下所示:
z;ld;snp;config;cred;log;n_samples
dataset1.z;dataset1.ld;dataset1.snp;dataset1.config;dataset1.cred;dataset1.log;5363
dataset2.z;dataset2.ld;dataset2.snp;dataset2.config;dataset2.cred;dataset2.log;5363Z 文件
dataset.z 文件是一个空格分隔的文本文件,每行包含一个 SNP 的 GWAS 汇总统计数据。它按以下顺序包含必需的列名。(或许就是GWAS summary statistics?)
rsid 列包含 SNP 标识符。标识符可以是 rsID 编号或染色体名称和基因组位置的组合(例如 XXX:yyy)。
chromosome列包含染色体名称。染色体名称可以通过预先计算的SNP相关性自由选择(例如“X”,“0X”或“chrX”)。
position列包含基本碱基对位置。
allele1列包含SNP的“第一个”等位基因。在SNPTEST中,对应“allele_A”,而BOLT-LMM使用“ALLELE1”
allele2列包含SNP的“第二个”等位基因。在SNPTEST中,这对应于“allele_B”,而BOLT-LMM使用“ALLELE0”
maf列包含次要等位基因频率
beta 列包含 GWAS 软件给出的估计效应量
se 列包含 GWAS 软件给出的效应大小的标准误差
flip可选列 - 见下文
fine-mapping需要beta列和se列,maf用于输出在allelic scale 上估计的后验效应。
使用 BCOR 时, rsid、染色体、位置、等位基因1 和等位基因 2 列中每个 SNP 的条目需要与 BCOR 文件中的信息相对应。染色体列可能必须包含 X = 1,...,9 的“0X”,其中 X 是染色体数。
建议根据其中一个等位基因的等位基因计数计算所有 SNP 相关性。在这种情况下,如果软件始终编码与效应等位基因相同的等位基因,则可以直接使用来自GWAS软件的估计效应大小及其标准误差。软件SNPTEST(使用“allele_B”作为效应等位基因)和BOLT-LMM(使用“ALLELE1”作为效应等位基因)就是这种情况。但是,如果GWAS软件将SNP的次要等位基因编码为效应等位基因,则需要将估计效应大小的方向翻转到第一个或第二个等位基因。这可以通过指定 --flip-beta 命令行参数并通过翻转列扩充 dataset.z 来完成,如果 SNP 的估计效应大小的方向需要翻转,则在行中包含 1,否则为 0。
SNP不必按基因组位置排序,可以驻留在不同的染色体上。但是,dataset.z 中 SNP 的顺序必须与 dataset.ld 中 SNP 的顺序相对应!
具有三个 SNP 的 dataset.z 文件可能如下所示。
rsid chromosome position allele1 allele2 maf beta se
rs1 10 1 T C 0.35 0.0050 0.0208
rs2 10 1 A G 0.04 0.0368 0.0761
rs3 10 1 G A 0.18 0.0228 0.0199LD文件
dataset.ld 文件是一个空格分隔的文本文件,包含 SNP 相关矩阵(皮尔逊相关)。
理想情况下,SNP相关矩阵是根据GWAS汇总统计数据所来自的相同样本上的基因型数据计算的。点这里看如果来自参考基因型(例如1000基因组计划)的SNP相关性与GWAS汇总统计数据不匹配,会发生什么情况。
使用imputed的生物样本库规模基因型数据,从GWAS软件中使用的相同基因型数据计算SNP相关性非常重要。阅读这里的示例,该示例强调了从GWAS软件中使用的相同剂量数据计算SNP相关性的重要性。例如,如果使用 BOLT-LMM 使用 SNP 剂量生成 GWAS 汇总统计数据(例如,当与 BGEN 文件一起使用时),则需要从相同的 SNP 剂量数据计算 SNP 相关性。这同样适用于使用-method预期选项处理基因型不确定性时的SNPTEST。如果使用BGEN文件从SNP剂量数据计算GWAS汇总统计数据,我们建议使用LDstore2软件计算SNP相关性,并且不建议将基因型概率转换为最佳猜测基因型以计算SNP相关性。
dataset.ld 中 SNP 的顺序必须与 dataset.z 中 SNP 的顺序相对应。
具有三个 SNP 的 dataset.ld 文件可能如下所示。
1.00 0.95 0.98
0.95 1.00 0.96
0.97 0.96 1.00BCOR文件
具体描述看这里。LD文件和BCOR文件只需要其中一种即可。
可选的K文件
默认情况下,FINEMAP假设SNP是因果关系,先验概率为1 /(基因组区域中SNP的数量)。作为替代方案,可以使用 dataset.k 文件指定基因组区域中因果 SNP 数量的先验概率。这是一个以空格分隔的文本文件,包含 k = 1,...,K 时的先验概率 pk = Pr(因果 SNPs 的次数为 k),其中 K 是 dataset.k 文件中的条目数。先前的概率必须是非负的,并且将被归一化为总和为1。
我们假设基因组区域至少包括一个因果SNP,因此p0 = 0。基因组区域中没有因果 SNP 的非零先验概率 p0 可以使用命令行参数 --prior-k0 指定。此值仅在计算后验概率 pk|data = Pr(因果 SNP 的次数为 k | 数据)时使用,但在fine-mapping本身期间不使用此值。我们进一步假设 pk = 0 表示 k = K + 1,...,m,其中 m 是 dataset.z 文件中的 SNP 数。
一个 dataset.k 文件允许三个 p1 = 0.6、p2 = 0.3 和 p3 = 0.1 的因果 SNP,如下所示。
0.6 0.3 0.1
输出说明
The output from FINEMAP is (1) a list of potential causal configurations together with their posterior probabilities and Bayes factors and, (2) for each variant, the posterior probability and Bayes factor of being causal.
SNP文件
dataset.snp 文件是一个空格分隔的文本文件。它包含每个 SNP 的 GWAS 汇总统计量和模型平均后验汇总,每行一个SNP。
index列包含 SNPs 在 dataset.z 文件中显示的行号。
rsid, chromosome, position, allele1 and allele2列是 Z 文件中的 SNP 标识符。
maf 列包含 Z 文件中给出的次要等位基因频率。
beta 列包含 Z 文件中给出的估计效应大小。
se 列包含 Z 文件中给出的效应大小估计值的标准误差。
z 列包含 z 分数。
prob列包含边际后验包含概率 (PIP)。第 i 个 SNP 的 PIP 是该 SNP 是因果关系的后验概率。
log10bf 列包含 log10 贝叶斯因子。贝叶斯因子量化了第 l 个 SNP 是因果关系的证据,log10 贝叶斯因子大于 2 表明了大量证据。
mean列包含与列beta中相同等位基因的后验效应大小均值的边际收缩估计值。第 l 个 SNP 的边际收缩估计值是通过从 dataset.config 文件中的所有因果configurations中平均此 SNP 的后验效应大小均值来计算的,假设如果因果配置中没有 SNP,则第 l 个 SNP 的效应大小为零。
sd 列包含后效应大小标准差的边际收缩估计值。这些估计值的计算方式与后效应大小均值的边际收缩估计值相同。
mean_incl列包含与列beta中相同的等位基因的后效应大小均值的条件估计值。第 l 个 SNP 的条件估计值是通过从config 文件中包含此 SNP 的因果configurations中平均此 SNP 的后验效应大小均值来计算的。
sd_incl列包含后验效应大小标准差的条件估计值。估计值的计算方式与后验效应大小均值的条件估计值相同。
prob列中的 PIP 是通过对包含 l 个 SNP 的 dataset.config 文件中所有因果从configurations的后验概率求和来计算的。如果允许的因果 SNP 的最大数量设置为 1,并且使用 --n-causal-snps 命令行参数,则 PIP 的总和为 1.0。
CONFIG文件
dataset.config 文件是一个以空格分隔的文本文件。每行包含每个因果configuration的后验summaries。
rank列包含排名。
config列包含 SNP 标识符。
prob列包含configurations是因果configurations的后验概率。
log10bf 列包含 log10 贝叶斯因子。贝叶斯因子量化了因果configurations优于空configurations的证据(没有SNP是因果关系)。
odds列包含顶级因果配置的odds
k 列包含top因果configurations的 SNP 数。
prob_norm_k列包含后验概率,即configurations是在具有相同数量 SNP 的configurations集上规范化的因果configurations。
h2 列包含 SNP 的遗传力贡献值。
h2_0.95CI 列包含 SNP 遗传力贡献的 95% 可信区间。
mean列包含joint后验效应大小均值。
sd 列包含joint后验效应大小标准差。
CRED 文件
dataset.cred 文件是一个以空格分隔的文本文件。它包含基因组区域中每个因果信号的95%可信集。对于每个可信的集合,提供了以下后验摘要:
每个 SNP 在可信集合中的后验概率
可信集合中 SNP 之间的最小、平均和中位数绝对相关性
log10 贝叶斯因子量化了除其他因果信号外还有因果信号的证据
CRED文件是为那些在基因组区域中具有最大后验概率的k个因果SNP的情况生成的。对于特定的k,FINEMAP采用具有最高后验概率的k-SNP因果configuration,然后询问该集合中的第l个SNP,这是可能在此因果configuration中取代该SNP的其他candidates。第 l 个可信集合显示最佳候选 SNP 及其在 k-SNP 因果configuration中的后验概率,该configuration还包含 k - 1 个 SNP。请注意,k - 1 个 SNP 被选为在其可信集合中具有最高的后验概率。
LOG文件
数据集.log文件输出其他信息,包含以下输出。
对于 k = 1,...,K,后验概率 pk|data = Pr(基因组区域中因果 SNP 的数量为 k |数据),其中 K 是允许的因果 SNP 的最大数量。
基因组区域中因果SNP的预期数量。
log10贝叶斯因子,用于量化基因组区域中至少一个因果SNP的证据。
模型平均遗传力和 95% 可信区间,以量化因果 SNP 的贡献

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









