






Tuning parameters of dimensionality reduction methods for single-cell RNA-seq analysis
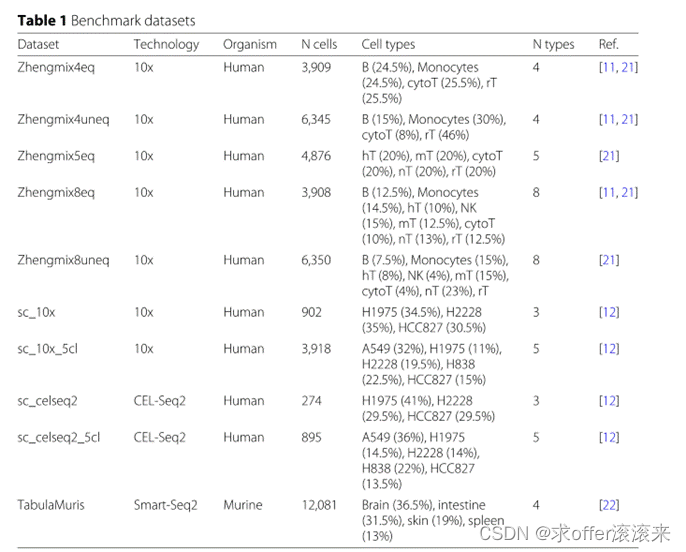
Datasets
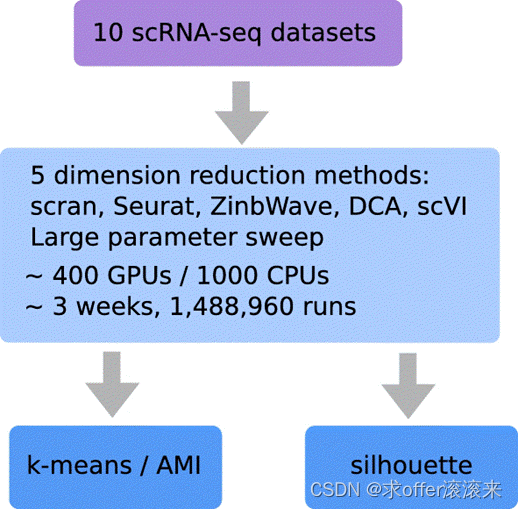
Method
Based on PCA
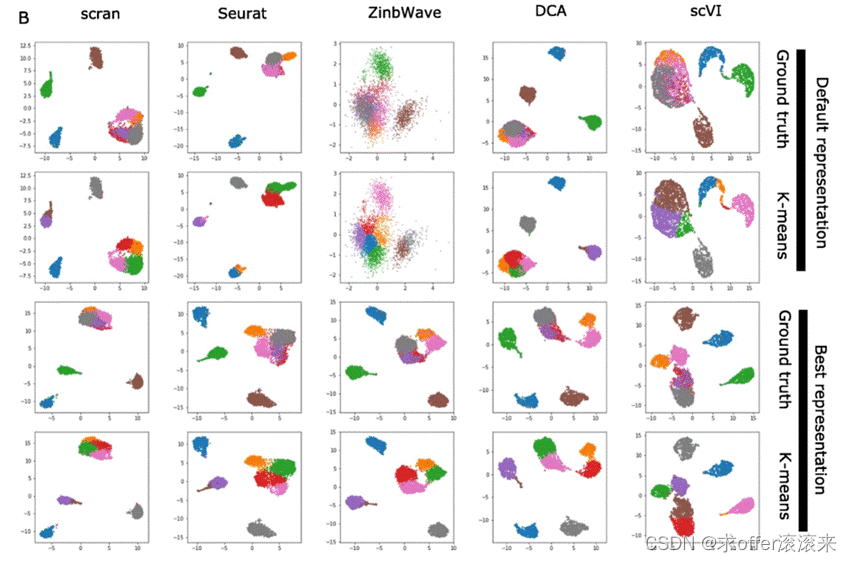
每种方法(列中)使用参数优化后(底部两行)的默认参数(顶部两行)表示 DR 后的 Zigmix8eq 的 UMAP 表示。在每行中,细胞根据其真实细胞类型(第 1 行和第 3 行)或基于 k 均值聚类进行着色。
- 1.scran:scran是一种基于主成分分析(PCA)的降维方法。它使用PCA来识别数据中最具变异性的主成分,然后将数据投影到这些主成分上,从而降低数据的维度。在默认参数下,scran在降维过程中表现竞争力,并且不太需要参数调整。
- 2.Seurat:Seurat也是一种基于PCA的降维方法。它使用PCA来提取数据中的主成分,并通过构建一个低维表示空间来呈现数据。Seurat是一种广泛使用的工具,具有丰富的功能和分析选项。
像 scran 这样简单的基于 PCA 的方法,即使使用默认参数,在 AMI 方面的参数调整后也足以匹配更复杂模型的性能;然而,一旦调整后,更复杂的模型的更好轮廓对于 k 均值聚类之外的其他下游应用程序可能是一个优势。进一步说明了参数调整的好处,该图显示了通过使用默认参数(底部两行)或 AMI 参数调整后(顶部两行)的不同方法学习的表示空间的 UMAP 可视化,对于zhenmix8eq 数据集。对于 ZinbWave 和 scVI,在这种情况下从参数调整中受益匪浅,我们看到数据集在默认参数或参数优化后看起来非常不同,不同的细胞类型在后一种情况下更好地分离。
Clustering
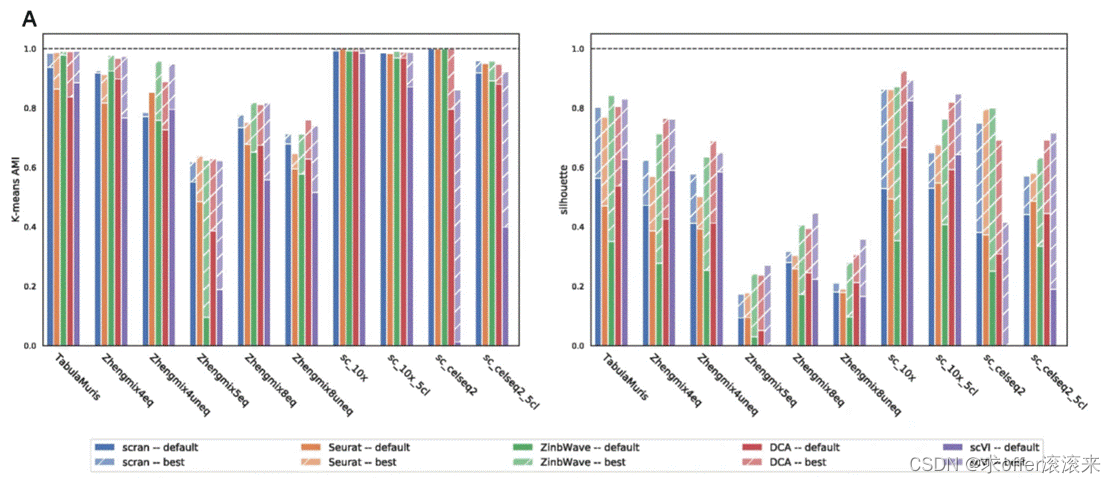
AMI是一种用于评估聚类算法性能的度量指标。它衡量了两个聚类结果之间的相似性,考虑了随机聚类的影响,并对聚类标签的不同命名提供了一定的容忍度。AMI的取值范围在0到1之间,值越接近1表示聚类结果越一致。
silhouette score是另一个常用的聚类评估指标,用于衡量聚类结果的紧密度和分离度。它结合了每个样本与其自身簇内的距离和与其他簇的距离,计算出一个介于-1到1之间的分数。silhouette score越接近1,表示样本在其所属簇内聚类得更紧密且与其他簇之间分离得更好。
在这篇文章中,AMI和silhouette score被用来评估不同维度降低方法在单细胞RNA测序数据中的性能。这些指标被用来衡量维度降低方法将相同细胞类型的细胞映射到表示空间中是否靠近彼此。
ARI是一种用于比较聚类结果之间的相似性的指标。它考虑了在随机分配聚类标签的情况下的预期分配,并度量了两个聚类结果之间的一致性程度。ARI的取值范围在-1到1之间,其中1表示完全一致的聚类结果,0表示与随机分配的聚类结果一致,-1表示完全不一致的聚类结果。
在这篇文章中,ARI被用来评估不同的维度降低方法在单细胞RNA测序数据中的性能。它用于衡量这些方法将相同细胞类型的细胞映射到表示空间中是否靠近彼此,并与已知的细胞类型标签进行比较,以评估聚类的准确性。
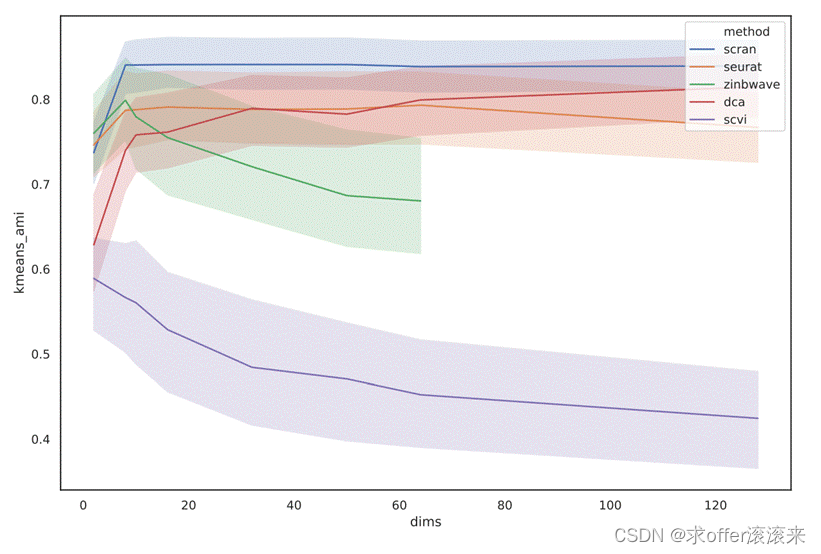
scran 的平均 AMI 最好(平均 AMI = 0.84),其次是 Seurat、ZinbWave 和 DCA(平均 AMI = 0.75∼0.79)
Conclusion
- 1.在单细胞RNA测序(scRNA-seq)数据的降维方法中,基于主成分分析(PCA)的方法(如scran和Seurat)在默认参数下具有竞争力,但对参数调整的效果不明显。
- 2.在需要参数调整的情况下,比较复杂的模型(如ZinbWave、DCA和scVI)可以达到更好的性能。
- 3.提出了一个基于十个具有已知细胞身份的scRNA-seq数据集的基准测试方案,通过系统地改变各种可调参数,评估了五种常用的降维方法的性能。
- 4.提供了两种自动调整参数的策略,可以通过更改默认参数或在每个新数据集上优化启发式方法来调整参数。
- 5.对于某些方法(如ZinbWave),这两种策略有时会识别出非常次优的参数,这表明在没有基准标注的复杂降维模型上进行参数调整仍然是一个重要但尚未解决的问题。
-
Canonical correlation analysis for multi-omics:Application to cross-cohort analysis
Datasets
- 1.TOPMed Informatics Research Center提供的数据集(3R01HL-117626-02S1; 合同HHSN268201800002I)
- 2.TOPMed Data Coordinating Center提供的核心支持,包括表型协调、数据管理、样本身份质量控制和程序协调(R01HL-120393; U01HL-120393; 合同HHSN268201800001I)
- 3.TOPMed MESA Multi-Omics项目(HHSN2682015000031/HSN26800004)
- 4.Jackson Heart Study(JHS)项目(HHSN268201300049C and HHSN268201300050C)
- 5.MESA和MESA SHARe项目
Canonical correlation analysis
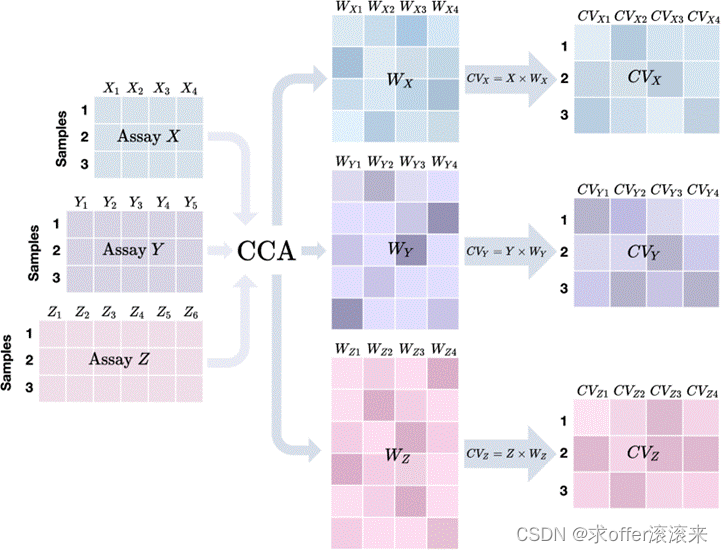
其中对三个样品进行了三种测定(X、Y 和 Z)。假设特征是连续的,没有分布假设。为了简洁起见,我们仅展示如何获取前 4 个简历。对于每个测定,CCA 推断 4 个权重向量(例如测定 X 的 WX1、WX2、WX3 和 WX4),从而产生四个 CV。例如,CVX1(测定 X 的顶部 CV)是通过 X×WX1 获得的。通过最大化三个测定中 CV 的相关性来推断权重。请注意,在最右边的 CV 矩阵中,CV 矩阵的每一列都是相应测定的一个 CV。此外,对应于相同列交叉测定的 CV 预计具有最大相关性(例如,CVX1、CVY1、CVZ1 相关性最强),而不同列中的 CV 预计在相同列中彼此正交或独立。
在典范相关分析(Canonical Correlation Analysis, CCA)中,典范变量(Canonical Variables)是通过对原始变量进行线性组合而得到的新变量。CCA旨在寻找两组变量之间的最大相关性,并通过典范变量来描述这种相关性。
对于第一组变量,典范变量是在该组变量中具有最高相关性和方差的线性组合。同样地,对于第二组变量,也存在与第一组典范变量最高相关性和方差的相应典范变量。
典范变量是CCA中的重要输出,它们可以用于解释两组变量之间的关系。通过分析典范变量和其对应的典范相关系数,可以揭示变量之间的模式和结构。
总结来说,典范变量是典范相关分析中通过线性组合原始变量而得到的新变量,用于描述两组变量之间的最大相关性。它们提供了一种方式来理解和解释多组变量之间的关系。
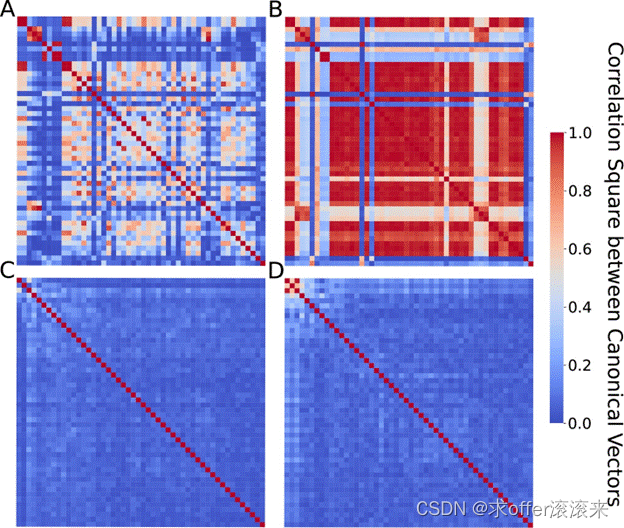
Regression analysis
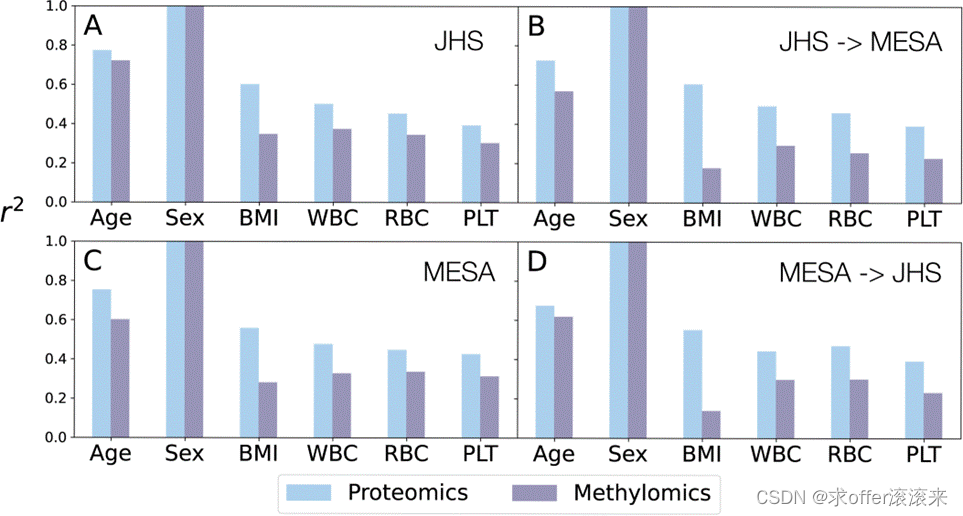
Conclusion
规范相关分析(CCA)可以应用于多组学数据,用于跨队列分析。CCA可以帮助整合来自不同组学平台的数据,并发现它们之间的共同模式和关系。同时,CCA也可以用于识别在多组学研究中不同队列中共有的生物标志物或特征。然而,需要注意CCA在多组学研究中可能存在的一些限制。















 1583
1583











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









