







网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
### 序列去冗余 Dereplicate
并添加miniuniqusize最小为8或1/1M,去除低丰度噪音并增加计算速度
-sizeout输出丰度, --relabel必须加序列前缀更规范, 1s
vsearch --derep_fulllength temp/filtered.fa
–minuniquesize 10 --sizeout --relabel Uni_
–output temp/uniques.fa
#高丰度非冗余序列非常小(500K~5M较适合),名称后有size和频率
### 聚类OTU/去噪ASV Cluster or denoise
#有两种方法:推荐unoise3去噪获得单碱基精度ASV,备选传统的97%聚类OTU(属水平精度)
#usearch两种特征挑选方法均自带de novo去嵌合体
#-minsize二次过滤,控制OTU/ASV数量至1-5千,方便下游统计分析
#方法1. 97%聚类OTU,适合大数据/ASV规律不明显/reviewer要求
#结果耗时1s, 产生508 OTUs, 去除126 chimeras
usearch -cluster_otus temp/uniques.fa -minsize 10
-otus temp/otus.fa
-relabel OTU_
#方法2. ASV去噪 Denoise: predict biological sequences and filter chimeras
#6s, 1530 good, 41 chimeras, 序列百万条可能需要几天/几周
usearch -unoise3 temp/uniques.fa -minsize 10
-zotus temp/zotus.fa
#修改序列名:Zotu为改为ASV方便识别
sed ‘s/Zotu/ASV_/g’ temp/zotus.fa > temp/otus.fa
查看otus.fa,序列名称都更改了。

### 基于参考去嵌合 Reference-based chimera detect
这里如果去除嵌合体的话就需要有rdp的数据库了,需提前下载好
不推荐,容易引起假阴性,因为参考数据库无丰度信息
而de novo时要求亲本丰度为嵌合体16倍以上防止假阴性
因为已知序列不会被去除,数据库选择越大越合理,假阴性率最低
mkdir -p result/raw
方法1. vsearch+rdp去嵌合(快但容易假阴性)
可自行下载silva并解压(替换rdp_16s_v18.fa为silva_16s_v123.fa),极慢但理论上更好
vsearch --uchime_ref temp/otus.fa
-db ${db}/usearch/rdp_16s_v18.fa
–nonchimeras result/raw/otus.fa
RDP: 7s, 143 (9.3%) chimeras; SILVA:9m, 151 (8.4%) chimeras
Win vsearch结果添加了windows换行符^M需删除,mac不要执行此命令,linux下应该也不需要
sed -i ‘s/\r//g’ result/raw/otus.fa
方法2. 不去嵌合,直接cp到指定工作目录
cp -f temp/otus.fa result/raw/otus.fa
### 特征表构建和筛选 Feature table create and filter
# OTU和ASV统称为特征(Feature),它们的区别是:
# OTU通常按97%聚类后挑选最高丰度或中心的代表性序列;
# ASV是基于序列进行去噪(排除或校正错误序列,并挑选丰度较高的可信序列)作为代表性序列
#### 生成特征表
id(1):100%相似度比对49.45%序列,1m50s
id(0.97):97%相似度比对83.66%序列,1m10s(更高数据使用率,更快)
time vsearch --usearch_global temp/filtered.fa
--db result/raw/otus.fa
--id 0.97 --threads 4
--otutabout result/raw/otutab.txt
#212862 of 268019 (79.42%)可比对
vsearch结果windows用户删除换行符^M校正为标准Linux格式
sed -i ‘s/\r//’ result/raw/otutab.txt
head -n6 result/raw/otutab.txt | cut -f 1-6 |cat -A
csvtk统计表行列
这里一定看好列数,是不是等于你的样品数;如果不等,一般是样品命名存在问题,具体看上面解释
csvtk -t stat result/raw/otutab.txt
#### 物种注释,且/或去除质体和非细菌 Remove plastid and non-Bacteria
先统计otu表的特征数,包括每个样品的otu总数
其他pipeline的筛选可根据需求和喜好来吧,不确定或感觉没必要的掠过即可,直接统计otu表信息
物种注释-去除质体和非细菌/古菌并统计比例(可选)
RDP物种注释(rdp_16s_v18)更快,但缺少完整真核来源数据,可能不完整,耗时15s;
SILVA数据库(silva_16s_v123.fa)更好注释真核、质体序列,极慢耗时3h起
置信阈值通常0.6/0.8,vserch最低0.1/usearch可选0输出最相似物种注释用于观察潜在分类
vsearch --sintax result/raw/otus.fa
–db ${db}/usearch/rdp_16s_v18.fa
–sintax_cutoff 0.1
–tabbedout result/raw/otus.sintax
sed -i ‘s/\r//’ result/raw/otus.sintax
########################
#物种注释结果:
ASV_7 d:Bacteria(1.00),p:Bacteroidetes(1.00),c:Flavobacteriia(1.00),o:Flavobacteriales(1.00),f:Flavobacteriaceae(1.00),g:Flavobacterium(1.00) + d:Bacteria,p:Bacteroidetes,c:Flavobacteriia,o:Flavobacteriales,f:Flavobacteriaceae,g:Flavobacterium
ASV_5 d:Bacteria(1.00),p:Cyanobacteria/Chloroplast(1.00),c:Chloroplast(1.00),f:Chloroplast(1.00),g:Streptophyta(1.00) + d:Bacteria,p:Cyanobacteria/Chloroplast,c:Chloroplast,f:Chloroplast,g:Streptophyta
ASV_4 d:Bacteria(1.00),p:Proteobacteria(1.00),c:Betaproteobacteria(1.00),o:Burkholderiales(1.00),f:Comamonadaceae(1.00),g:Rhizobacter(0.98) + d:Bacteria,p:Proteobacteria,c:Betaproteobacteria,o:Burkholderiales,f:Comamonadaceae,g:Rhizobacter
ASV_3 d:Bacteria(1.00),p:Proteobacteria(1.00),c:Betaproteobacteria(1.00),o:Burkholderiales(1.00),f:Comamonadaceae(1.00),g:Rhizobacter(0.93) + d:Bacteria,p:Proteobacteria,c:Betaproteobacteria,o:Burkholderiales,f:Comamonadaceae,g:Rhizobacter
ASV_2 d:Bacteria(1.00),p:Proteobacteria(1.00),c:Betaproteobacteria(1.00),o:Burkholderiales(1.00),f:Comamonadaceae(1.00),g:Pelomonas(1.00) + d:Bacteria,p:Proteobacteria,c:Betaproteobacteria,o:Burkholderiales,f:Comamonadaceae,g:Pelomonas
#OTU表简单统计 Summary OTUs table
usearch -otutab_stats result/otutab.txt
-output result/otutab.stat
cat result/otutab.stat
207572 Reads (207.6k)
18 Samples
1363 OTUs
24534 Counts
4507 Count =0 (18.4%)
4229 Count =1 (17.2%)
3999 Count >=10 (16.3%)
413 OTUs found in all samples (30.3%)
576 OTUs found in 90% of samples (42.3%)
1276 OTUs found in 50% of samples (93.6%)
Sample sizes: min 10425, lo 10953, med 11549, mean 11531.8, hi 11924, max 13209
#### 等量抽样标准化 Normlize by subsample
这里主要是依赖R包了,使用R运行,可以脚本运行,或者直接将otutab\_rare.R文件打开在R终端或studio的交互窗口直接运行
#使用vegan包进行等量重抽样,输入reads count格式Feature表result/otutab.txt
#可指定输入文件、抽样量和随机数,输出抽平表result/otutab_rare.txt和多样性alpha/vegan.txt
mkdir -p result/alpha
Rscript ${db}/script/otutab_rare.R --input result/otutab.txt
--depth 10000 --seed 1
--normalize result/otutab_rare.txt
--output result/alpha/vegan.txt
#再统计查看抽样后的信息
usearch -otutab_stats result/otutab_rare.txt
-output result/otutab_rare.stat
cat result/otutab_rare.stat
#### 计算α多样性 calculate alpha diversity
使用USEARCH计算14种alpha多样性指数(Chao1有错勿用)
#details in http://www.drive5.com/usearch/manual/alpha_metrics.html
usearch -alpha_div result/otutab_rare.txt
-output result/alpha/alpha.txt
#当然大家可使用自己的R程序来进行alpha多样性的计算
#### 计算稀释丰富度 calculate rarefaction richness
#稀释曲线:取1%-100%的序列中OTUs数量,每次无放回抽样
#Rarefaction from 1%, 2% … 100% in richness (observed OTUs)-method without_replacement https://drive5.com/usearch/manual/cmd_otutab_subsample.html
usearch -alpha_div_rare result/otutab_rare.txt
-output result/alpha/alpha_rare.txt
-method without_replacement
#预览结果
head -n2 result/alpha/alpha_rare.txt
#样本测序量低出现非数值"-"的处理,详见常见问题8
sed -i “s/-/\t0.0/g” result/alpha/alpha_rare.txt
#### 筛选高丰度菌 Filter by abundance
#计算各特征的均值,有组再求分组均值,需根据实验设计metadata.txt修改组列名
#输入文件为feautre表result/otutab.txt,实验设计metadata.txt
#输出为特征表按组的均值-一个实验可能有多种分组方式
#-h显示脚本帮助(参数说明)
Rscript ${db}/script/otu_mean.R -h
#scale是否标准化,zoom标准化总和,all输出全部样本均值,type计算类型mean或sum
Rscript ${db}/script/otu_mean.R --input result/otutab.txt
--metadata result/metadata.txt
--group Group --thre 0
--scale TRUE --zoom 100 --all TRUE --type mean
--output result/otutab_mean.txt
结果为全部和各组均值
head -n3 result/otutab_mean.txt
#如以平均丰度>0.1%筛选,可选0.5或0.05,得到每个组的OTU组合
awk ‘BEGIN{OFS=FS=“\t”}{if(FNR==1) {for(i=3;i<=NF;i++) a[i]=KaTeX parse error: Expected 'EOF', got '}' at position 24: … "OTU","Group";}̲ \ else {fo…i>0.1) print $1, a[i];}}’
result/otutab_mean.txt > result/alpha/otu_group_exist.txt
head result/alpha/otu_group_exist.txt
cut -f 2 result/alpha/otu_group_exist.txt | sort | uniq -c
试一试:不同丰度下各组有多少OTU/ASV
可在 http://ehbio.com/test/venn/ 中绘图并显示各组共有和特有维恩或网络图
也可在 http://www.ehbio.com/ImageGP 绘制Venn、upSetView和Sanky
#### β多样性 Beta diversity
#结果有多个文件,需要目录
mkdir -p result/beta/
#基于OTU构建进化树 Make OTU tree, 4s
usearch -cluster_agg result/otus.fa -treeout result/otus.tree
#生成5种距离矩阵:bray_curtis, euclidean, jaccard, manhatten, unifrac
usearch -beta_div result/otutab_rare.txt -tree result/otus.tree
-filename_prefix result/beta/
#### 物种注释分类汇总
#OTU对应物种注释2列格式:去除sintax中置信值,只保留物种注释,替换:为_,删除引号
cut -f 1,4 result/otus.sintax
|sed ‘s/\td/\tk/;s/😕__/g;s/,/;/g;s/"//g’
> result/taxonomy2.txt
head -n3 result/taxonomy2.txt
#OTU对应物种8列格式:注意注释是非整齐
#生成物种表格OTU/ASV中空白补齐为Unassigned
awk ‘BEGIN{OFS=FS=“\t”}{delete a; a[“k”]=“Unassigned”;a[“p”]=“Unassigned”;a[“c”]=“Unassigned”;a[“o”]=“Unassigned”;a[“f”]=“Unassigned”;a[“g”]=“Unassigned”;a[“s”]=“Unassigned”;
split($2,x,“;”);for(i in x){split(x[i],b,"");a[b[1]]=b[2];}
print $1,a[“k”],a[“p”],a[“c”],a[“o”],a[“f”],a[“g”],a[“s”];}’
result/taxonomy2.txt > temp/otus.tax
sed 's/;/\t/g;s/.//g;’ temp/otus.tax|cut -f 1-8 |
sed ‘1 s/^/OTUID\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/’
> result/taxonomy.txt
head -n3 result/taxonomy.txt
#统计门纲目科属,使用 rank参数 p c o f g,为phylum, class, order, family, genus缩写
mkdir -p result/tax
for i in p c o f g;do
usearch -sintax_summary result/otus.sintax
-otutabin result/otutab_rare.txt -rank KaTeX parse error: Expected group after '_' at position 31: … result/tax/sum_̲{i}.txt
done
sed -i ‘s/(//g;s/)//g;s/"//g;s/#//g;s//Chloroplast//g’ result/tax/sum_*.txt
列出所有文件
wc -l result/tax/sum_*.txt
head -n3 result/tax/sum_g.txt
#### 有参定量特征表
比对Greengenes97% OTUs比对,用于PICRUSt/Bugbase功能预测
mkdir -p result/gg/
usearch比对更快,但文件超限报错选附录14 vsearch比对
默认10核以下使用1核,10核以上使用10核
usearch -otutab temp/filtered.fa -otus ${db}/gg/97_otus.fa
-otutabout result/gg/otutab.txt -threads 4
比对率80.0%, 1核11m,4核3m,10核2m,内存使用743Mb
head -n3 result/gg/otutab.txt
#统计
usearch -otutab_stats result/gg/otutab.txt -output result/gg/otutab.stat
cat result/gg/otutab.stat
#### 空间清理及数据提交
这个步骤应该放在最后文章撰写完成后再处理,因为有些中间文件可能需要调整参数重新处理,有些大文件不用再重复cp了,另外,提交数据也是在文章接收后才会用到。
#删除中间大文件
rm -rf temp/*.fq
分双端统计md5值,用于数据提交
cd seq
md5sum _1.fq.gz > md5sum1.txt
md5sum _2.fq.gz > md5sum2.txt
paste md5sum1.txt md5sum2.txt | awk ‘{print $2"\t"$1"\t"$4"\t"$3}’ | sed 's///g’ > …/result/md5sum.txt
rm md5sum
cd …
cat result/md5sum.txt
#### 统计分析及出图
##### Alpha多样性
#Alpha多样性箱线图,对于不同样品数的作图,建议单个运行后再逐个调整参数以使出图布局最佳
查看帮助
Rscript ${db}/script/alpha_boxplot.R -h
完整参数,多样性指数可选richness chao1 ACE shannon simpson invsimpson
Rscript ${db}/script/alpha_boxplot.R --alpha_index richness
--input result/alpha/vegan.txt --design result/metadata.txt
--group Group --output result/alpha/
--width 89 --height 59
使用循环绘制6种常用指数
for i in head -n1 result/alpha/vegan.txt|cut -f 2-;do
Rscript ${db}/script/alpha_boxplot.R --alpha_index ${i}
--input result/alpha/vegan.txt --design result/metadata.txt
--group Group --output result/alpha/
--width 89 --height 59
done
mv alpha_boxplot_TukeyHSD.txt result/alpha/
Alpha多样性柱状图+标准差
Rscript ${db}/script/alpha_barplot.R --alpha_index richness
--input result/alpha/vegan.txt --design result/metadata.txt
--group Group --output result/alpha/
--width 89 --height 59
1.2 稀释曲线
Rscript ${db}/script/alpha_rare_curve.R
--input result/alpha/alpha_rare.txt --design result/metadata.txt
--group Group --output result/alpha/
--width 120 --height 59
1.3 多样性维恩图
三组比较:-f输入文件,-a/b/c/d/g分组名,-w/u为宽高英寸,-p输出文件名后缀
bash ${db}/script/sp_vennDiagram.sh
-f result/alpha/otu_group_exist.txt
-a WT -b KO -c OE
-w 3 -u 3
-p WT_KO_OE
四组比较,图和代码见输入文件目录,运行目录为当前项目根目录
bash ${db}/script/sp_vennDiagram.sh
-f result/alpha/otu_group_exist.txt
-a WT -b KO -c OE -d All
-w 3 -u 3
-p WT_KO_OE_All
EVenn在线绘制维恩图 https://www.ehbio.com/test/venn
##### Beta多样性
2.1 距离矩阵热图pheatmap
添加分组注释,如2,4列的基因型和地点
cut -f 1-2 result/metadata.txt > temp/group.txt
以bray_curtis为例,-f输入文件,-h是否聚类TRUE/FALSE,-u/v为宽高英寸
-P添加行注释文件,-Q添加列注释
bash ${db}/script/sp_pheatmap.sh
-f result/beta/bray_curtis.txt
-H ‘TRUE’ -u 6.9 -v 5.6
-P temp/group.txt -Q temp/group.txt
##### 主坐标分析PCoA
输入文件,选择分组,输出文件,图片尺寸mm,统计见beta_pcoa_stat.txt
Rscript ${db}/script/beta_pcoa.R
--input result/beta/bray_curtis.txt --design result/metadata.txt
--group Group --label FALSE --width 89 --height 59
--output result/beta/bray_curtis.pcoa.pdf
添加样本标签 --label TRUE
Rscript ${db}/script/beta_pcoa.R
--input result/beta/bray_curtis.txt --design result/metadata.txt
--group Group --label TRUE --width 89 --height 59
--output result/beta/bray_curtis.pcoa.label.pdf
mv beta_pcoa_stat.txt result/beta/
##### 限制性主坐标分析CPCoA
Rscript ${db}/script/beta_cpcoa.R
--input result/beta/bray_curtis.txt --design result/metadata.txt
--group Group --output result/beta/bray_curtis.cpcoa.pdf
--width 89 --height 59
添加样本标签 --label TRUE
Rscript ${db}/script/beta_cpcoa.R
--input result/beta/bray_curtis.txt --design result/metadata.txt
--group Group --label TRUE --width 89 --height 59
--output result/beta/bray_curtis.cpcoa.label.pdf
##### 物种组成Taxonomy
3.1 堆叠柱状图Stackplot
以门§水平为例,结果包括output.sample/group.pdf两个文件
Rscript ${db}/script/tax_stackplot.R
--input result/tax/sum_p.txt --design result/metadata.txt
--group Group --color ggplot --legend 7 --width 89 --height 59
--output result/tax/sum_p.stackplot
修改颜色–color ggplot, manual1(22), Paired(12) or Set3(12)
Rscript ${db}/script/tax_stackplot.R
--input result/tax/sum_p.txt --design result/metadata.txt
--group Group --color Paired --legend 12 --width 181 --height 119
--output result/tax/sum_p.stackplotPaired
批量绘制输入包括p/c/o/f/g共5级
for i in p c o f g; do
Rscript KaTeX parse error: Expected group after '_' at position 55: … result/tax/sum_̲{i}.txt --design result/metadata.txt
--group Group --output result/tax/sum_${i}.stackplot
--legend 8 --width 89 --height 59; done
3.2 弦/圈图circlize
以纲(class,c)为例,绘制前5组
i=c
Rscript KaTeX parse error: Expected group after '_' at position 54: … result/tax/sum_̲{i}.txt --design result/metadata.txt
--group Group --legend 5
结果位于当前目录circlize.pdf(随机颜色),circlize_legend.pdf(指定颜色+图例)
移动并改名与分类级一致
mv circlize.pdf result/tax/sum_KaTeX parse error: Expected group after '_' at position 55: … result/tax/sum_̲{i}.circlize_legend.pdf
3.3 树图treemap(参考)
多层级包含物种关系,输入特征表和物种注释,输出树图
指定包含特征数量和图片宽高,100个ASV耗时12s
Rscript ${db}/script/tax_maptree.R
--input result/otutab.txt --taxonomy result/taxonomy.txt
--output result/tax/tax_maptree.pdf
--topN 100 --width 183 --height 118
##### 差异比较
1. R语言差异分析
1.1 差异比较
Error in file(file, ifelse(append, “a”, “w”)),输出目录不存在,创建目录即可
mkdir -p result/compare/
输入特征表、元数据;指定分组列名、比较组和丰度
选择方法 wilcox/t.test/edgeR、pvalue和fdr和输出目录
compare=“KO-WT”
Rscript ${db}/script/compare.R
--input result/otutab.txt --design result/metadata.txt
--group Group --compare ${compare} --threshold 0.1
--method edgeR --pvalue 0.05 --fdr 0.2
--output result/compare/
1.2 火山图
输入compare.R的结果,输出火山图带数据标签,可指定图片大小
Rscript
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
v
o
l
c
a
n
o
.
R
−
−
i
n
p
u
t
r
e
s
u
l
t
/
c
o
m
p
a
r
e
/
{db}/script/compare_volcano.R \ --input result/compare/
db/script/comparevolcano.R −−inputresult/compare/{compare}.txt
--output result/compare/${compare}.volcano.pdf
--width 89 --height 59
1.3 热图
输入compare.R的结果,筛选列数,指定元数据和分组、物种注释,图大小英寸和字号
bash
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
h
e
a
t
m
a
p
.
s
h
−
i
r
e
s
u
l
t
/
c
o
m
p
a
r
e
/
{db}/script/compare_heatmap.sh -i result/compare/
db/script/compareheatmap.sh−iresult/compare/{compare}.txt -l 7
-d result/metadata.txt -A Group
-t result/taxonomy.txt
-w 8 -h 5 -s 7
-o result/compare/${compare}
1.4 曼哈顿图
i差异比较结果,t物种注释,p图例,w宽,v高,s字号,l图例最大值
图例显示不图,可增加高度v为119+即可,后期用AI拼图为KO-WT.heatmap.emf
bash
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
m
a
n
h
a
t
t
a
n
.
s
h
−
i
r
e
s
u
l
t
/
c
o
m
p
a
r
e
/
{db}/script/compare_manhattan.sh -i result/compare/
db/script/comparemanhattan.sh−iresult/compare/{compare}.txt
-t result/taxonomy.txt
-p result/tax/sum_p.txt
-w 183 -v 59 -s 7 -l 10
-o result/compare/${compare}.manhattan.p.pdf
上图只有6个门,切换为纲c和-L Class展示细节
bash
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
m
a
n
h
a
t
t
a
n
.
s
h
−
i
r
e
s
u
l
t
/
c
o
m
p
a
r
e
/
{db}/script/compare_manhattan.sh -i result/compare/
db/script/comparemanhattan.sh−iresult/compare/{compare}.txt
-t result/taxonomy.txt
-p result/tax/sum_c.txt
-w 183 -v 59 -s 7 -l 10 -L Class
-o result/compare/${compare}.manhattan.c.pdf
显示完整图例,再用AI拼图
bash
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
m
a
n
h
a
t
t
a
n
.
s
h
−
i
r
e
s
u
l
t
/
c
o
m
p
a
r
e
/
{db}/script/compare_manhattan.sh -i result/compare/
db/script/comparemanhattan.sh−iresult/compare/{compare}.txt
-t result/taxonomy.txt
-p result/tax/sum_c.txt
-w 183 -v 149 -s 7 -l 10 -L Class
-o result/compare/${compare}.manhattan.c.legend.pdf
1.5 单个特征的绘制
筛选显示差异ASV,按KO组丰度降序列,取ID展示前10
awk ‘$4<0.05’ result/compare/KO-WT.txt | sort -k7,7nr | cut -f1 | head
差异OTU细节展示
Rscript ${db}/script/alpha_boxplot.R --alpha_index ASV_2
--input result/otutab.txt --design result/metadata.txt
--transpose TRUE --scale TRUE
--width 89 --height 59
--group Group --output result/compare/feature_
ID不存在会报错: Error in data.frame(…, check.names = FALSE) : 参数值意味着不同的行数: 0, 18 Calls: alpha_boxplot -> cbind -> cbind -> data.frame
指定某列排序:按属丰度均值All降序
csvtk -t sort -k All:nr result/tax/sum_g.txt | head
差属细节展示
Rscript ${db}/script/alpha_boxplot.R --alpha_index Lysobacter
--input result/tax/sum_g.txt --design result/metadata.txt
--transpose TRUE
--width 89 --height 59
--group Group --output result/compare/feature_
1.5 三元图
#参考示例见:result\compare\ternary\ternary.Rmd 文档
#备选教程246.三元图的应用与绘图实战
##### STAMP输入文件准备
2.1 生成输入文件
Rscript ${db}/script/format2stamp.R -h
mkdir -p result/stamp
Rscript ${db}/script/format2stamp.R --input result/otutab.txt
--taxonomy result/taxonomy.txt --threshold 0.01
--output result/stamp/tax
可选Rmd文档见result/format2stamp.Rmd
2.2 绘制扩展柱状图和表
compare=“KO-WT”
替换ASV(result/otutab.txt)为属(result/tax/sum_g.txt)
Rscript ${db}/script/compare_stamp.R
--input result/stamp/tax_5Family.txt --metadata result/metadata.txt
--group Group --compare
c
o
m
p
a
r
e
−
−
t
h
r
e
s
h
o
l
d
0.1
−
−
m
e
t
h
o
d
"
t
.
t
e
s
t
"
−
−
p
v
a
l
u
e
0.05
−
−
f
d
r
"
n
o
n
e
"
−
−
w
i
d
t
h
189
−
−
h
e
i
g
h
t
159
−
−
o
u
t
p
u
t
r
e
s
u
l
t
/
s
t
a
m
p
/
{compare} --threshold 0.1 \ --method "t.test" --pvalue 0.05 --fdr "none" \ --width 189 --height 159 \ --output result/stamp/
compare−−threshold0.1 −−method"t.test"−−pvalue0.05−−fdr"none" −−width189−−height159 −−outputresult/stamp/{compare}
可选Rmd文档见result/CompareStamp.Rmd
##### LEfSe输入文件准备
3.1. 命令行生成文件
可选命令行生成输入文件
Rscript ${db}/script/format2lefse.R -h
mkdir -p result/lefse
threshold控制丰度筛选以控制作图中的枝数量
Rscript ${db}/script/format2lefse.R --input result/otutab.txt
--taxonomy result/taxonomy.txt --design result/metadata.txt
--group Group --threshold 0.4
--output result/lefse/LEfSe
3.2 Rmd生成输入文件(可选)
#1. result目录中存在otutab.txt, metadata.txt, taxonomy.txt三个文件;
#2. Rstudio打开EasyAmplicon中format2lefse.Rmd,另存至result目录并Knit生成输入文件和可重复计算网页;
3.3 LEfSe分析
#方法1. 打开LEfSe.txt并在线提交 https://www.bic.ac.cn/BIC/#/analysis?page=b%27MzY%3D%27
#方法2. LEfSe本地分析(限Linux系统、选学),参考代码见附录
#方法3. LEfSe官网在线使用
#### QIIME 2分析流程
代码详见 qiime2/pipeline\_qiime2.sh
##### 功能预测
## 1. PICRUSt功能预测
### PICRUSt 1.0
方法1. 使用 http://www.ehbio.com/ImageGP 在线分析 gg/otutab.txt
方法2. Linux服务器用户可参考"附录2. PICRUSt功能预测"实现软件安装和分析
然后结果使用STAMP/R进行差异比较
R语言绘图
输入文件格式调整
l=L2
sed ‘/# Const/d;s/OTU //’ result/picrust/all_level.ko.
l
.
t
x
t
>
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{l}.txt > result/picrust/
l.txt>result/picrust/{l}.txt
num=head -n1 result/picrust/${l}.txt|wc -w
paste <(cut -f
n
u
m
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
num result/picrust/
numresult/picrust/{l}.txt) <(cut -f 1-
[
n
u
m
−
1
]
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
[num-1] result/picrust/
[num−1]result/picrust/{l}.txt)
> result/picrust/
l
.
s
p
f
c
u
t
−
f
2
−
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{l}.spf cut -f 2- result/picrust/
l.spfcut−f2−result/picrust/{l}.spf > result/picrust/${l}.mat.txt
awk 'BEGIN{FS=OFS=“\t”} {print $2,KaTeX parse error: Expected 'EOF', got '}' at position 2: 1}̲' result/picrus…{l}.spf | sed ‘s/;/\t/’ | sed ‘1 s/ID/Pathway\tCategory/’
> result/picrust/${l}.anno.txt
差异比较
compare=“KO-WT”
Rscript
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
.
R
−
−
i
n
p
u
t
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{db}/script/compare.R \ --input result/picrust/
db/script/compare.R −−inputresult/picrust/{l}.mat.txt --design result/metadata.txt
--group Group --compare ${compare} --threshold 0
--method wilcox --pvalue 0.05 --fdr 0.2
--output result/picrust/
可对结果${compare}.txt筛选
绘制指定组(A/B)的柱状图,按高分类级着色和分面
Rscript
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
h
i
e
r
a
r
c
h
y
f
a
c
e
t
.
R
−
−
i
n
p
u
t
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{db}/script/compare_hierarchy_facet.R \ --input result/picrust/
db/script/comparehierarchyfacet.R −−inputresult/picrust/{compare}.txt
--data MeanA
--annotation result/picrust/
l
.
a
n
n
o
.
t
x
t
−
−
o
u
t
p
u
t
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{l}.anno.txt \ --output result/picrust/
l.anno.txt −−outputresult/picrust/{compare}.MeanA.bar.pdf
绘制两组显著差异柱状图,按高分类级分面
Rscript
d
b
/
s
c
r
i
p
t
/
c
o
m
p
a
r
e
h
i
e
r
a
r
c
h
y
f
a
c
e
t
2.
R
−
−
i
n
p
u
t
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{db}/script/compare_hierarchy_facet2.R \ --input result/picrust/
db/script/comparehierarchyfacet2.R −−inputresult/picrust/{compare}.txt
--pvalue 0.05 --fdr 0.1
--annotation result/picrust/
l
.
a
n
n
o
.
t
x
t
−
−
o
u
t
p
u
t
r
e
s
u
l
t
/
p
i
c
r
u
s
t
/
{l}.anno.txt \ --output result/picrust/
l.anno.txt −−outputresult/picrust/{compare}.bar.pdf
### PICRUSt 2.0
软件安装见附录6. PICRUSt环境导出和导入
(可选)PICRUSt2(Linux/Windows下Linux子系统,要求>16GB内存)
安装参考附录5的方式直接下载安装包并解压即可使用
Linux中加载conda环境
conda activate picrust2
进入工作目录,服务器要修改工作目录
wd=/mnt/d/amplicon/result/picrust2
mkdir -p ${wd} && cd ${wd}
运行流程,内存15.7GB,耗时12m
picrust2_pipeline.py -s …/otus.fa -i …/otutab.txt -o ./out -p 8
添加EC/KO/Pathway注释
cd out
add_descriptions.py -i pathways_out/path_abun_unstrat.tsv.gz -m METACYC
-o METACYC.tsv
add_descriptions.py -i EC_metagenome_out/pred_metagenome_unstrat.tsv.gz -m EC
-o EC.tsv
add_descriptions.py -i KO_metagenome_out/pred_metagenome_unstrat.tsv.gz -m KO
-o KO.tsv
KEGG按层级合并
db=/mnt/d/EasyMicrobiome/
python3 ${db}/script/summarizeAbundance.py
-i KO.tsv
-m ${db}/kegg/KO1-4.txt
-c 2,3,4 -s ‘,+,+,’ -n raw
-o KEGG
统计各层级特征数量
wc -l KEGG*
可视化见picrust2文件夹中ggpicrust2.Rmd
##### 元素循环FAPROTAX
方法1. 在线分析,推荐使用 http://www.bic.ac.cn/ImageGP/index.php/Home/Index/FAPROTAX.html 在线分析
方法2. Linux下分析、如QIIME 2环境下
设置工作目录
wd=/mnt/d/amplicon/result/faprotax/
mkdir -p ${wd} && cd ${wd}
设置脚本目录
sd=/mnt/d/EasyMicrobiome/script/FAPROTAX_1.2.7
1. 软件安装
注:软件已经下载至 EasyMicrobiome/script目录,在qiime2环境下运行可满足依赖关系
#(可选)下载软件新版本,以1.2.7版为例, 2023/7/14更新数据库
#wget -c https://pages.uoregon.edu/slouca/LoucaLab/archive/FAPROTAX/SECTION_Download/MODULE_Downloads/CLASS_Latest%20release/UNIT_FAPROTAX_1.2.7/FAPROTAX_1.2.7.zip
#解压
#unzip FAPROTAX_1.2.7.zip
#新建一个python3环境并配置依赖关系,或进入qiime2 python3环境
conda activate qiime2-2023.7
source /home/silico_biotech/miniconda3/envs/qiime2/bin/activate
#测试是否可运行,弹出帮助即正常工作
python $sd/collapse_table.py
2. 制作输入OTU表
#txt转换为biom json格式
biom convert -i …/otutab_rare.txt -o otutab_rare.biom --table-type=“OTU table” --to-json
#添加物种注释
biom add-metadata -i otutab_rare.biom --observation-metadata-fp …/taxonomy2.txt
-o otutab_rare_tax.biom --sc-separated taxonomy
--observation-header OTUID,taxonomy
#指定输入文件、物种注释、输出文件、注释列名、属性列名
3. FAPROTAX功能预测
#python运行collapse_table.py脚本、输入带有物种注释OTU表tax.biom、
#-g指定数据库位置,物种注释列名,输出过程信息,强制覆盖结果,结果文件和细节
#下载faprotax.txt,配合实验设计可进行统计分析
#faprotax_report.txt查看每个类别中具体来源哪些OTUs
python ${sd}/collapse_table.py -i otutab_rare_tax.biom
-g ${sd}/FAPROTAX.txt
--collapse_by_metadata ‘taxonomy’ -v --force
-o faprotax.txt -r faprotax_report.txt
4. 制作OTU对应功能注释有无矩阵
对ASV(OTU)注释行,及前一行标题进行筛选
grep ‘ASV_’ -B 1 faprotax_report.txt | grep -v -P ‘^–$’ > faprotax_report.clean
faprotax_report_sum.pl脚本将数据整理为表格,位于public/scrit中
perl ${sd}/…/faprotax_report_sum.pl -i faprotax_report.clean -o faprotax_report
查看功能有无矩阵,-S不换行
less -S faprotax_report.mat
##### Bugbase细菌表型预测
1. Bugbase命令行分析
cd
w
d
/
r
e
s
u
l
t
b
u
g
b
a
s
e
=
{wd}/result bugbase=
wd/resultbugbase={db}/script/BugBase
rm -rf bugbase/
脚本已经优化适合R4.0,biom包更新为biomformat
Rscript ${bugbase}/bin/run.bugbase.r -L ${bugbase}
-i gg/otutab.txt -m metadata.txt -c Group -o bugbase/
2. 其它可用分析
使用 http://www.bic.ac.cn/ImageGP/index.php/Home/Index/BugBase.html
官网,https://bugbase.cs.umn.edu/ ,有报错,不推荐
Bugbase细菌表型预测Linux,详见附录3. Bugbase细菌表型预测
##### MachineLearning机器学习
RandomForest包使用的R代码见advanced/RandomForestClassification和RandomForestRegression
Silme2随机森林/Adaboost使用代码见EasyMicrobiome/script/slime2目录中的slime2.py,详见附录4
使用实战(使用QIIME 2的Python3环境,以在Windows中为例)
conda activate qiime2-2023.7
cd /mnt/d/EasyMicrobiome/script/slime2
#使用adaboost计算10000次(16.7s),推荐千万次
./slime2.py otutab.txt design.txt --normalize --tag ab_e4 ab -n 10000
#使用RandomForest计算10000次(14.5s),推荐百万次,支持多线程
./slime2.py otutab.txt design.txt --normalize --tag rf_e4 rf -n 10000
#### Evolution进化树
cd ${wd}
mkdir -p result/tree
cd ${wd}/result/tree
1. 筛选高丰度/指定的特征
#方法1. 按丰度筛选特征,一般选0.001或0.005,且OTU数量在30-150个范围内
#统计特征表中ASV数量,如总计1609个
tail -n+2 …/otutab_rare.txt | wc -l
#按相对丰度0.2%筛选高丰度OTU
usearch -otutab_trim …/otutab_rare.txt
-min_otu_freq 0.002
-output otutab.txt
#统计筛选OTU表特征数量,总计~81个
tail -n+2 otutab.txt | wc -l
#方法2. 按数量筛选
#按丰度排序,默认由大到小
usearch -otutab_sortotus …/otutab_rare.txt \
-output otutab_sort.txt
#提取高丰度中指定Top数量的OTU ID,如Top100,
sed ‘1 s/#OTU ID/OTUID/’ otutab_sort.txt \
| head -n101 > otutab.txt
#修改特征ID列名
sed -i ‘1 s/#OTU ID/OTUID/’ otutab.txt
#提取ID用于提取序列
cut -f 1 otutab.txt > otutab_high.id
筛选高丰度菌/指定差异菌对应OTU序列
usearch -fastx_getseqs …/otus.fa -labels otutab_high.id
-fastaout otus.fa
head -n 2 otus.fa
筛选OTU对物种注释
awk ‘NR==FNR{a[$1]=$0} NR>FNR{print a[$1]}’ …/taxonomy.txt
otutab_high.id > otutab_high.tax
#获得OTU对应组均值,用于样本热图
#依赖之前otu_mean.R计算过按Group分组的均值
awk ‘NR==FNR{a[$1]=$0} NR>FNR{print a[$1]}’ …/otutab_mean.txt otutab_high.id
| sed ‘s/#OTU ID/OTUID/’ > otutab_high.mean
head -n3 otutab_high.mean
#合并物种注释和丰度为注释文件
cut -f 2- otutab_high.mean > temp
paste otutab_high.tax temp > annotation.txt
head -n 3 annotation.txt
2. 构建进化树
起始文件为 result/tree目录中 otus.fa(序列)、annotation.txt(物种和相对丰度)文件
Muscle软件进行序列对齐,3s
muscle -in otus.fa -out otus_aligned.fas
方法1. 利用IQ-TREE快速构建ML进化树,2m
rm -rf iqtree
mkdir -p iqtree
iqtree -s otus_aligned.fas
-bb 1000 -redo -alrt 1000 -nt AUTO
-pre iqtree/otus
方法2. FastTree快速建树(Linux)
注意FastTree软件输入文件为fasta格式的文件,而不是通常用的Phylip格式。输出文件是Newick格式。
该方法适合于大数据,例如几百个OTUs的系统发育树!
Ubuntu上安装fasttree可以使用apt install fasttree
fasttree -gtr -nt otus_aligned.fas > otus.nwk
3. 进化树美化
访问http://itol.embl.de/,上传otus.nwk,再拖拽下方生成的注释方案于树上即美化
方案1. 外圈颜色、形状分类和丰度方案
annotation.txt OTU对应物种注释和丰度,
-a 找不到输入列将终止运行(默认不执行)-c 将整数列转换为factor或具有小数点的数字,-t 偏离提示标签时转换ID列,-w 颜色带,区域宽度等, -D输出目录,-i OTU列名,-l OTU显示名称如种/属/科名,
cd ${wd}/result/tree
Rscript ${db}/script/table2itol.R -a -c double -D plan1 -i OTUID -l Genus -t %s -w 0.5 annotation.txt
生成注释文件中每列为单独一个文件
方案2. 生成丰度柱形图注释文件
Rscript ${db}/script/table2itol.R -a -d -c none -D plan2 -b Phylum -i OTUID -l Genus -t %s -w 0.5 annotation.txt
方案3. 生成热图注释文件
Rscript ${db}/script/table2itol.R -c keep -D plan3 -i OTUID -t %s otutab.txt
方案4. 将整数转化成因子生成注释文件
Rscript ${db}/script/table2itol.R -a -c factor -D plan4 -i OTUID -l Genus -t %s -w 0 annotation.txt
树iqtree/otus.contree在 http://itol.embl.de/ 上展示,拖拽不同Plan中的文件添加树注释
返回工作目录
cd ${wd}
4. 进化树可视化
https://www.bic.ac.cn/BIC/#/ 提供了更简易的可视化方式
附加视频
目录 Supp,网课有对应视频(可能编号不同,找关键字)
S1. 网络分析R/CytoGephi
目录 Supp/S1NetWork
S2. 溯源和马尔可夫链
目录 Supp/S2SourcetrackerFeastMarkov
S11、网络分析ggClusterNet
代码:advanced/ggClusterNet/Practice.Rmd
S12、Microeco包数据可视化
代码:advanced/microeco/Practice.Rmd
## 附录:Linux服务器下分析(选学)
#注:Windows下可能无法运行以下代码,推荐在Linux,或Windows下Linux子系统下conda安装相关程序
1. LEfSe分析
mkdir -p ~/amplicon/lefse
cd ~/amplicon/lefse
format2lefse.Rmd代码制作或上传输入文件LEfSe.txt
安装lefse
conda install lefse
#格式转换为lefse内部格式
lefse-format_input.py LEfSe.txt input.in -c 1 -o 1000000
#运行lefse
run_lefse.py input.in input.res
#绘制物种树注释差异
lefse-plot_cladogram.py input.res cladogram.pdf --format pdf
#绘制所有差异features柱状图
lefse-plot_res.py input.res res.pdf --format pdf
#绘制单个features柱状图(同STAMP中barplot)
head input.res #查看差异features列表
lefse-plot_features.py -f one --feature_name “Bacteria.Firmicutes.Bacilli.Bacillales.Planococcaceae.Paenisporosarcina”
--format pdf input.in input.res Bacilli.pdf
#批量绘制所有差异features柱状图,慎用(几百张差异结果柱状图阅读也很困难)
mkdir -p features
lefse-plot_features.py -f diff --archive none --format pdf
input.in input.res features/
2. PICRUSt功能预测
#推荐使用 http://www.bic.ac.cn/BIC/#/analysis?tool_type=tool&page=b%27Mzk%3D%27 在线分析
#有Linux服务器用户可参考以下代码搭建本地流程
n=picrust
conda create -n ${n} ${n} -c bioconda -y
wd=/mnt/d/amplicon
cd $wd/result/gg
启动环境
conda activate picrust
#上传gg/otutab.txt至当前目录
#转换为OTU表通用格式,方便下游分析和统计
biom convert -i otutab.txt
-o otutab.biom
--table-type=“OTU table” --to-json
设置数据库目录,如 /mnt/d/db
db=~/db
#校正拷贝数,30s, 102M
normalize_by_copy_number.py -i otutab.biom
-o otutab_norm.biom
-c ${db}/picrust/16S_13_5_precalculated.tab.gz
#预测宏基因组KO表,3m,1.5G,biom方便下游归类,txt方便查看分析
predict_metagenomes.py -i otutab_norm.biom
-o ko.biom
-c ${db}/picrust/ko_13_5_precalculated.tab.gz
predict_metagenomes.py -f -i otutab_norm.biom
-o ko.txt
-c ${db}/picrust/ko_13_5_precalculated.tab.gz
#按功能级别分类汇总, -c输出KEGG_Pathways,分1-3级
sed -i ‘/# Constru/d;s/#OTU //’ ko.txt
num=head -n1 ko.txt|wc -w
paste <(cut -f
n
u
m
k
o
.
t
x
t
)
<
(
c
u
t
−
f
1
−
num ko.txt) <(cut -f 1-
numko.txt)<(cut−f1−[num-1] ko.txt) > ko.spf
for i in 1 2 3;do
categorize_by_function.py -f -i ko.biom -c KEGG_Pathways -l
i
−
o
p
a
t
h
w
a
y
{i} -o pathway
i−opathway{i}.txt
sed -i ‘/# Const/d;s/#OTU //’ pathway${i}.txt
paste <(cut -f
n
u
m
p
a
t
h
w
a
y
num pathway
numpathway{i}.txt) <(cut -f 1-
[
n
u
m
−
1
]
p
a
t
h
w
a
y
[num-1] pathway
[num−1]pathway{i}.txt) > pathway${i}.spf
done
wc -l *.spf
3. Bugbase细菌表型预测
1. 软件安装(己整合到EasyMicrobiome中,原代码需要更新才能在当前运行)
#有两种方法可选,推荐第一种,可选第二种,仅需运行一次
#方法1. git下载,需要有git
git clone https://github.com/knights-lab/BugBase
#方法2. 下载并解压
wget -c https://github.com/knights-lab/BugBase/archive/master.zip
mv master.zip BugBase.zip
unzip BugBase.zip
mv BugBase-master/ BugBase
cd BugBase
#安装依赖包
export BUGBASE_PATH=pwd
export PATH=$PATH:pwd/bin
#安装了所有依赖包
run.bugbase.r -h
#测试数据
run.bugbase.r -i doc/data/HMP_s15.txt -m doc/data/HMP_map.txt -c HMPBODYSUBSITE -o output
2. 准备输入文件
cd ~/amplicon/result
#输入文件:基于greengene OTU表的biom格式(本地分析支持txt格式无需转换)和mapping file(metadata.txt首行添加#)
#上传实验设计+刚才生成的otutab_gg.txt
#生成在线分析使用的biom1.0格式
biom convert -i gg/otutab.txt -o otutab_gg.biom --table-type=“OTU table” --to-json
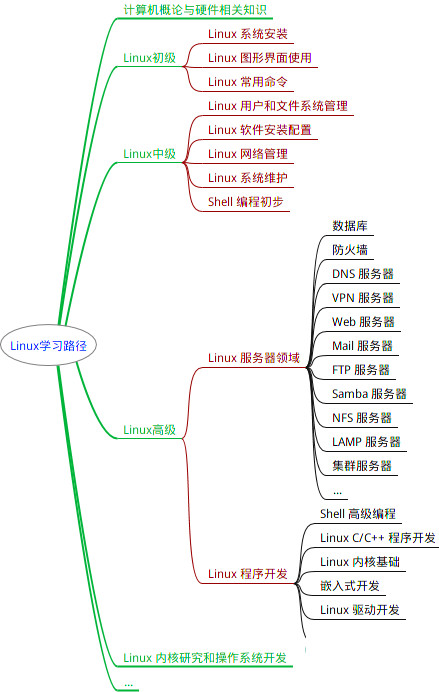
最全的Linux教程,Linux从入门到精通
======================
-
linux从入门到精通(第2版)
-
Linux系统移植
-
Linux驱动开发入门与实战
-
LINUX 系统移植 第2版
-
Linux开源网络全栈详解 从DPDK到OpenFlow

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!















 8439
8439

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









