







最后的话
最近很多小伙伴找我要Linux学习资料,于是我翻箱倒柜,整理了一些优质资源,涵盖视频、电子书、PPT等共享给大家!
资料预览
给大家整理的视频资料:
给大家整理的电子书资料:

如果本文对你有帮助,欢迎点赞、收藏、转发给朋友,让我有持续创作的动力!
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
-t 20 \
sample1 \
sample1.contigs_coverage.out \
sample1.contig.reads.sorted.bam
prodigal
cp sample1.nucle_seq.fa sample1
checkm coverage
-x fasta
-m 20
-t 20
sample1
sample1.gene_coverage.out
sample1.gene.reads.sorted.bam
结果包含contig序列ID、所在的bin的ID、coverage等信息,如下所示,用excel对齐了看吧:

* **Sequence Id:** 序列的唯一标识符。
* **Bin Id:** 该序列所属的Bin(即被分组到哪个类别)。在宏基因组学中,Bin通常指的是组装后相似特征或物种的组集合。
* **Sequence length (bp):** 序列的长度,以碱基对(bp)计算。
* **Bam Id:** 对应该序列的测序数据文件。
* **Coverage:** 覆盖度,指的是这个contig在样品中出现的平均次数。它通常由测序reads的比对情况得出。
* **Mapped reads:** 映射的reads数量,指的是能够成功比对到这个contig上的测序reads数量。
相对丰度计算公式:
要计算基因在样品中的相对丰度,您可以使用覆盖度和Mapped reads。通常情况下,丰度可以用覆盖度和测序reads的总数来估算。例如,可以使用以下公式计算相对丰度:
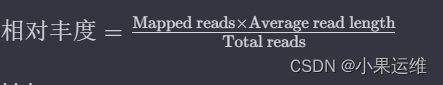
其中:
* Mapped reads 是该contig的映射reads数量。
* Average read length 是测序reads的平均长度。
* Total reads 是所有测序reads的总数。
Total reads统计,双端序列使用一个正向或反向的序列就行:
python脚本:
def count_reads_fastq(fastq_file):
with open(fastq_file, ‘r’) as f:
count = sum(1 for line in f) // 4 # 每4行代表一个read,因此除以4得到reads数量
return count
替换为您的FASTQ文件路径
file_path = ‘path/to/your/fastq_file.fastq’
调用函数计算reads数量
reads_count = count_reads_fastq(file_path)
print(f"FASTQ文件中的reads数量为: {reads_count}")
bash脚本:
统计FASTQ文件中reads的数量
grep -c “^@” your_fastq_file.fastq
这里需要注意第一列的sequence id,后续需要mapping到基因注释结果中对应的seq id,除此外,我们只需要reads的mapping信息即可。接下来可以根据map的reads数计算相对丰度,也即除以contig长度和总得reads数,类似于RNA-seq中的RPKM标准化方法。假如是多样品混合拼接,只需要将每一个样品的reads数据重复上面操作,最后根据contig id进行整合。
## 第二种方式:BWA(推荐)、SAMtools、CheckM
#首先对参考序列构建index:
bwa index -p sample1_gene sample1.nucle_seq.fa
#使用BWA-MEM进行比对:
bwa mem
-t 20
sample1_gene
sample1_clean_1.fastq
sample1_clean_2.fastq
>sample1_gene.sam
**从这里开始与第一种方式samtools步骤开始相同**
## 第三种方式: bedtools计算
步骤1:比对测序reads到参考基因组
假设使用Bowtie2进行比对
bowtie2-build your_genome.fa your_genome_index # 如果尚未构建索引
bowtie2 -x your_genome_index -U your_reads.fastq -S aligned.sam
步骤2:将比对结果转换为BAM格式
samtools view -S -b aligned.sam > aligned.bam
再sort排序一下
samtools sort aligned.bam -o aligned.sorted.bam --threads 20
samtools index aligned.sorted.bam
步骤3:提取覆盖度信息
假设使用bedtools进行提取覆盖度信息
bedtools genomecov -ibam aligned.sorted.bam > coverage.bed
步骤4:计算基因长度
假设已经有基因长度信息的文件,如genes_lengths.txt
步骤5:计算相对丰度
awk ‘BEGIN {OFS=“\t”} NR==FNR {len[$1]=$2; next} {print $1, $2/len[$1]}’ genes_coverage.txt genes_lengths.txt > relative_abundance.txt
## 第四种方式 salmon
Salmon是一个用于RNA-Seq数据分析的工具,用于估计基因表达水平。Salmon的优势在于其高效的计算速度和准确的表达量估计。它使用一种快速和有效的索引结构来对转录组进行建模,从而能够在较短的时间内处理大规模的RNA-Seq数据。
**尽管salmon工具主要是用于转录组序列的分析,但其计算序列或基因丰度的方法在宏基因组学中也可以得已应用**
使用Salmon进行基因表达分析的步骤如下:
1、建立索引:使用Salmon的"index"命令来建立索引。在建立索引时,您需要为参考基因组的序列文件和转录本注释文件提供相关信息。例如,以下命令可用于建立索引:
salmon index -t transcripts.fa -i index
示例: 一般fasta文件与reads应都为核酸序列,-i输出到指定文件夹
salmon index -t nucle.fa -i index
构建完索引后输出文件夹会包括以下文件:
duplicate_clusters.tsv hash.bin header.json indexing.log quasi_index.log refInfo.json rsd.bin sa.bin txpInfo.bin versionInfo.json
其中,"transcripts.fa"是转录本序列文件,"index"是输出索引文件的目录。
2、运行Salmon:使用Salmon的"quant"命令来运行表达量估计。在运行时,您需要提供输入的RNA-Seq数据文件、参考基因组的索引文件以及一些其他参数。例如,以下命令可用于运行Salmon:
salmon quant -i index -l A -r reads.fq -o output_dir
-i 以上面步骤输出的文件夹为输入, -1,-2分别为正反reads,-o 为输出文件夹,建议与前面索引文件夹独立
salmon quant
-i $PWD/index
-l IU
-1 $PWD/reads_1.fastq
-2 $PWD/freads_2.fastq
–validateMappings
-o quant.out
ls quant.out
aux_info cmd_info.json lib_format_counts.json libParams logs quant.sf
head quant.sf
Name Length EffectiveLength TPM NumReads
k141_859451_length_39317_cov_9.1141_1 468 139.968 13.816795 27.000
k141_859451_length_39317_cov_9.1141_2 2544 2211.374 4.081127 126.000
k141_859451_length_39317_cov_9.1141_3 699 366.406 3.323210 17.000
k141_859451_length_39317_cov_9.1141_4 537 205.175 4.189182 12.000
k141_859451_length_39317_cov_9.1141_5 705 372.406 3.846670 20.000
k141_859451_length_39317_cov_9.1141_6 1437 1104.374 4.345548 67.002
k141_859451_length_39317_cov_9.1141_7 1695 1362.374 3.364770 64.000
k141_859451_length_39317_cov_9.1141_8 279 35.120 6.118389 3.000
k141_859451_length_39317_cov_9.1141_9 1584 1251.374 5.952755 104.000
其中,"index"是之前建立的索引文件目录,"reads.fq"是RNA-Seq数据的FASTQ文件,"output\_dir"是输出结果的目录。
解读结果:Salmon的输出结果包括每个基因的表达量估计值和其他一些统计信息。您可以使用其他工具(如R或Python)来进一步分析和可视化这些表达量数据。
这只是Salmon工具的基本使用方法,还有许多其他参数和功能可以用于不同的分析需求。您可以查阅Salmon的官方文档以获取更详细的信息和使用示例。
## 全流程计算脚本
多样品请自己做for循环操作
### 自动分析脚本1
用于计算基于 `bwa-mem`、`samtools` 和 `CheckM` 的Contigs相对丰度。该脚本假设你已经有参考基因组和测序数据,并安装了相应的软件。
#!/bin/bash
定义文件路径
reference_genome=“your_reference_genome.fa”
reads=“your_reads.fastq”
步骤1:用bwa-mem比对测序reads到参考基因组
bwa index $reference_genome # 如果尚未构建索引
bwa mem -t 4 $reference_genome $reads > aligned.sam
步骤2:将比对结果转换为BAM格式
samtools view -bS aligned.sam > aligned.bam
samtools sort -o aligned_sorted.bam aligned.bam
samtools index aligned_sorted.bam
步骤3:使用CheckM估算Contigs的丰度
checkm lineage_wf -f checkm_output.txt -x fa $reference_genome contigs_dir/ checkm_results/
步骤4:提取覆盖度信息
checkm qa -o 2 -f checkm_coverage.txt checkm_results/lineage.ms contigs_dir/ coverage.txt
### 自动分析脚本2
基于`bowtie2`、`samtools`和`bedtools`计算Contigs或Genes在样品中相对丰度的流程自动分析脚本。请注意,这个脚本仅供参考,并不能直接运行,因为其中的文件路径、参数和具体数据可能需要根据实际情况进行调整。
#!/bin/bash
假设有参考基因组文件your_genome.fa,测序reads文件your_reads.fastq和基因注释文件genes_annotation.gff
步骤1:构建参考基因组索引
bowtie2-build your_genome.fa your_genome_index
步骤2:将测序reads比对到参考基因组
bowtie2 -x your_genome_index -U your_reads.fastq -S aligned.sam
步骤3:将比对结果转换为BAM格式
samtools view -S -b aligned.sam > aligned.bam
步骤4:生成基因覆盖度文件
bedtools genomecov -ibam aligned.bam -g your_genome.fa.fai > coverage.txt
步骤5:根据基因注释文件提取基因长度信息
awk ‘{if($3==“gene”) print $0}’ genes_annotation.gff | cut -f 1,4,5 > genes_lengths.txt
步骤6:根据覆盖度和基因长度信息计算相对丰度
awk ‘BEGIN {OFS=“\t”} NR==FNR {len[$1]=$3-$2; next} {print $1, $2/len[$1]}’ coverage.txt genes_lengths.txt > relative_abundance.txt
输出结果
echo “相对丰度计算完成。结果保存在 relative_abundance.txt 文件中。”
### 自动分析脚本3
python
import subprocess
import os
定义文件路径
ref_genome = ‘path/to/your_reference_genome.fasta’
sample_reads = ‘path/to/your_sample_reads.fastq’
gene_lengths = ‘path/to/your_gene_lengths.txt’
步骤1:比对测序reads到参考基因组
使用Bowtie2进行比对
bowtie_index = ‘your_genome_index’
subprocess.run([‘bowtie2-build’, ref_genome, bowtie_index])
subprocess.run([‘bowtie2’, ‘-x’, bowtie_index, ‘-U’, sample_reads, ‘-S’, ‘aligned.sam’])
步骤2:将比对结果转换为BAM格式
subprocess.run([‘samtools’, ‘view’, ‘-S’, ‘-b’, ‘aligned.sam’, ‘-o’, ‘aligned.bam’])
步骤3:提取覆盖度信息
subprocess.run([‘bedtools’, ‘genomecov’, ‘-ibam’, ‘aligned.bam’, ‘-g’, ref_genome + ‘.fai’, ‘>’, ‘coverage.bed’])
步骤4:计算基因长度
假设已经有基因长度信息的文件
步骤5:计算相对丰度
with open(‘genes_coverage.txt’, ‘r’) as cov_file, open(gene_lengths, ‘r’) as len_file, open(‘relative_abundance.txt’, ‘w’) as output:
for cov_line, len_line in zip(cov_file, len_file):
contig_id, coverage = cov_line.strip().split(‘\t’)
gene_id, length = len_line.strip().split(‘\t’)
rel_abundance = float(coverage) / float(length)
output.write(f"{gene_id}\t{rel_abundance}\n")
清理临时文件(可选)
os.remove(‘aligned.sam’)
os.remove(‘aligned.bam’)
os.remove(‘coverage.bed’)
###
### 自动分析脚本4
NGless 是一个用于分析宏基因组数据的领域特定语言(DSL)。
安装和使用参考:[生物信息学分析领域领先的特制语言环境NGLess(Next Generation Less)介绍、安装配置和详细使用方法-CSDN博客]( )
以下是一个使用 NGless 的示例脚本,用于计算 contigs 或 genes 在样品中的相对丰度。请注意,这只是一个简化的示例,实际的分析脚本可能需要根据具体的数据和需求进行调整。
加载所需模块
ngless “0.11”
最全的Linux教程,Linux从入门到精通
======================
-
linux从入门到精通(第2版)
-
Linux系统移植
-
Linux驱动开发入门与实战
-
LINUX 系统移植 第2版
-
Linux开源网络全栈详解 从DPDK到OpenFlow

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!















 965
965











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









