






背景
分子建模(Molecular modelling),或称分子模拟,指利用理论方法与计算技术,模拟出化学分子的外观或性质,是计算化学、材料科学和生物学的核心支柱[1]。
分子建模的理想方式是量子化学方法(quantum chemistry),包括从头算(ab initio)、 密度泛函理论(density functional theory)等,这种方法考虑了原子内部的量子特性,准确度高,但是计算成本也极高,仅可用于较小的系统。较大尺度下的建模方法常用分子动力学(molecular dynamics)方式,其对势能和原子力的预测必须既准确又有效,这样才能准确地描述复杂系统在长时间尺度上的演化。经典的立场方法,只使用基于的原子坐标的简单函数预测分子势能,在准确性上有所不足。而用机器学习模型来预测原子势能(machine learning interatomic potentials,MLIPs),有望以较高的计算效率提供准确的预测。其中,一类基于信息传递的神经网络模型在准确性表现上尤为突出。
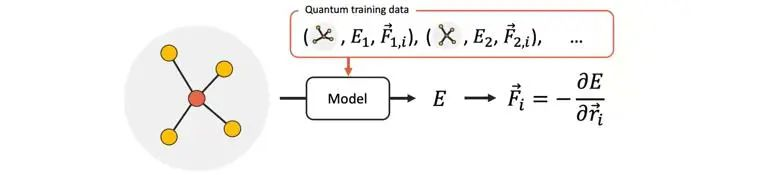
图1 神经网络模型预测分子势能
消息传递是图神经网络里的概念,在图神经网络中,我们希望相邻节点或边交换信息并借此更新各自的embedding。消息传递包含三个步骤(如图2所示)[2]:
1). 对图中的每个节点或边,收集所有相邻节点或边的embedding(或消息)。
2). 通过聚合函数(如求和)聚合所有消息。
3). 所有聚合的消息都通过一个更新函数更新目标节点或边(通常是一个神经网络)。
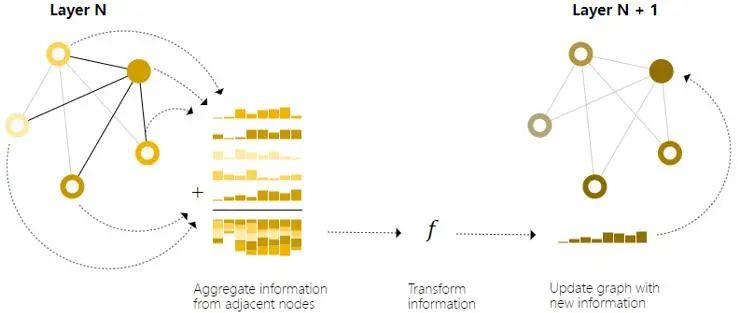
图2 GNN中的消息传递
经过多次消息传递后,初始图中的局域的性质可以影响到远处的节点,这样的现象又叫做图上的卷积操作。基于多次消息传递后所形成的新的图,可以实施预测任务[3]。
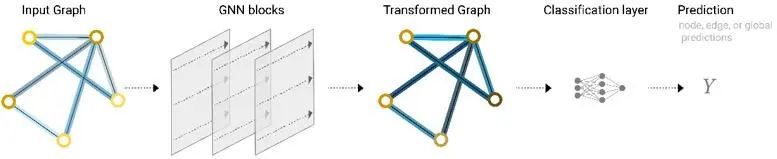
图3 GNN的完整工作流程
在分子势能预测领域,几何图神经网络(Geometric GNN),通过利用分子的几何信息(如距离、角度和二面角等),并且保持物理量在运算过程中的对称性(平移、旋转和反演),也得到了快速发展[4]。
其中,等变网络获得了极大关注,以NequIP[5]为例,由于引入等变性,其对数据集规模的需求大大降低。
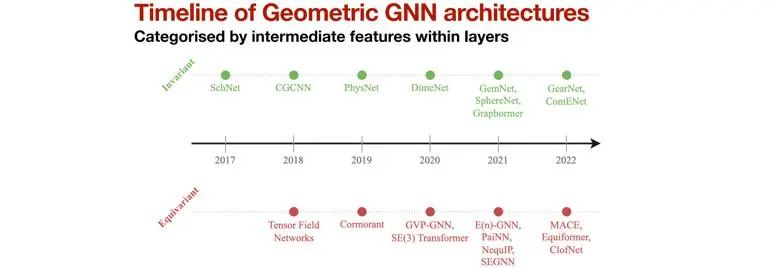
图4 Geometric GNN的发展
1、Allegro的网络结构
Allegro[6]是一种等变神经网络。关于等变性的相关概念,如不可约表示、张量积等,可以参考我们之前发布的知乎文章:MindSpore AI科学计算系列:等变神经网络与e3nn数学库。
相比于传统的基于消息传递的方法,Allegro主要有两处不同:使用有序原子对标记隐层特征、使用标量与张量的双通道架构取代消息传递机制。其整体网络结构如图5所示。
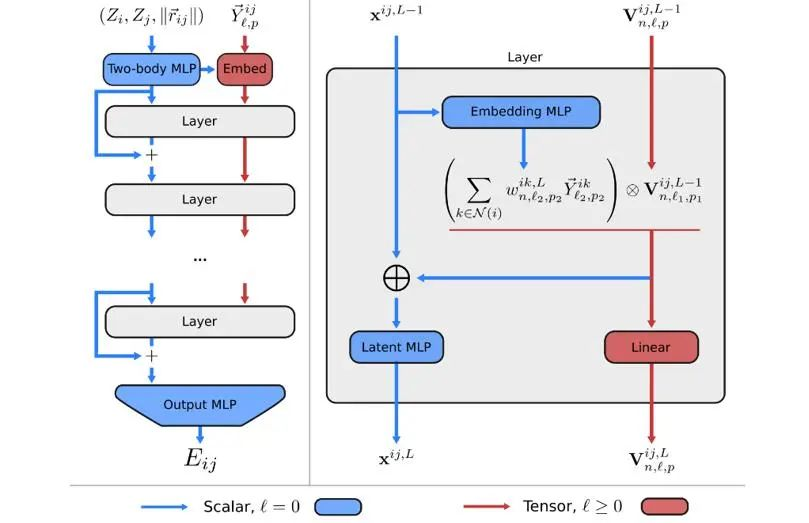
图5 Allegro网络结构
1.1 有序原子对标记隐层特征
Allegro中的隐层特征,是由有序原子对(有向边)(i,j)标记的,i表示中心原子。其信息更新也是由边与边的张量积完成。
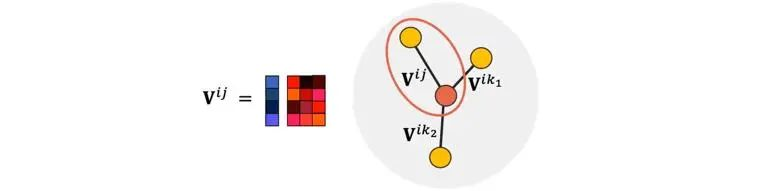
图6 Allegro中的有序原子对
1.2 使用标量与张量的双通道架构
图5中,蓝色通道表示不变的隐层空间,x^{ij}代表了保持不变特性的隐层特征(标量部分,l=0),对该部分可以使用任意操作。红色通道表示等变的隐层空间,V^{ij}代表了保持等变特性的隐层特征(张量部分,l>0),对该部分特征的操作需要遵循等变性。初始的隐层标量特征x^{ij}由原子种类和原子间距离的径向基投影(radial basis)拼接而成。
![]()
式1
初始的隐层张量特征V^{ij}为中心原子到邻域原子的距离矢量的球谐投影(spherical harmonics)的线性映射
![]()
式2
相比于标量运算,等变空间的张量运算需要消耗更大的算力。设计双通道的目的,即是希望用较小的计算代价来获得等变特征的信息。
具体地,对于每一层(layer),其由4部分组成:
1). 隐层标量特征通过MLP(Embedding MLP),嵌入中心原子的邻域环境信息,作为每一对边与边张量积的权重。
2). 边与边做张量积运算,在中心原子维度加权求和。
3). 取张量积运算结果(irreps)的标量通道输出的部分(l=0,p=1),拼接原隐层标量特征后,经过MLP(Latent MLP),更新x^{ij}。
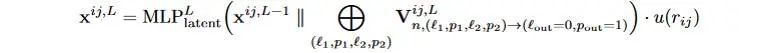
式3
4). 对张量积运算结果做等变线性层操作,更新V^{ij}。
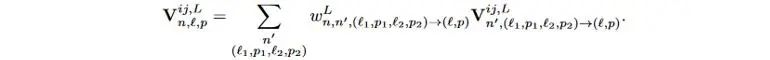
式4
其中,借助张量积运算的双线性特性,使得对于每一个中心原子的每条有向边,只需要做一次张量积运算,如下式所示。
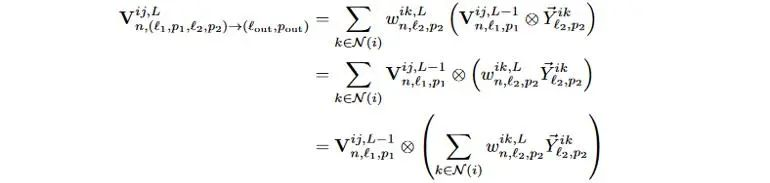
式5
在层与层之间的标量空间还引入了残差(residual),以便信息传播。
1.3 能量分解
一个系统的分子势能可以分解为系统中所有原子能量的总和:E_system=Σ(i)E_i。Allegro进一步将每个原子的能量分解为以该原子为中心与其邻域原子组成的边的能量:
E_i=Σ(j∈neighbor(i))E_{ij}。
因此,Allegro网络的输出即为E_{ij},可用于计算系统总能量。
2、结果
1. 预测准确性方面,Allegro在revMD-17数据集上取得了和NequIP相近的准确性,在QM9数据集上取得了最佳的准确性。
2. 由于Allegro的严格局部特性,作者还展示了其在并行计算下的性能优势。
3、感想
基于群表示理论的相关操作(不可约表示与张量积等),计算代价较高,提升计算性能是几何图神经网络的重要课题。传统的基于消息传递机制的几何图神经网络无法高效利用并行计算。Allegro不依赖于消息传递机制,保证了严格局部特性,从而能较好地利用并行计算。当前昇思MindSpore AI框架结合自身优势,基于已有能力开展Allegro相关工作,预计不久将上线MindSpore Chemistry。当分子系统规模较大时,能否高效利用并行计算尤为重要,这也是研究者们在设计模型之初应当首先考虑的问题之一。
参考文献
[1]Keith T Butler, Daniel W Davies, Hugh Cartwright, Olexandr Isayev, and Aron Walsh. 2018. Machine learning for molecular and materials science. Nature 559, 7715 (2018), 547–555.
[2]Tian Xie and Jeffrey C Grossman. 2018. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Physical Review Letters 120, 14 (2018), 145301.
[3]Keqiang Yan, Yi Liu, Yuchao Lin, and Shuiwang Ji. 2022. Periodic Graph Transformers for Crystal Material Property Prediction. In The 36th Annual Conference on Neural Information Processing Systems. 15066–15080.
往期回顾
MindSpore AI科学计算系列 | 周期性图Transformer提升MindSpore模型对晶体性质的预测
MindSpore AI科学计算系列 | 初探海洋大模型AI-GOMS,打开大模型在海洋方面的大门















 4万+
4万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









