






论文标题:A deep learning framework for in silico screening of anticancer drugs at the single-cell level

思维导图:
目录
1. 文献速览
肿瘤异质性在肿瘤进展和对临床治疗的耐药性中起着关键作用。单细胞RNA测序能够探索细胞群体中的异质性,识别稀有细胞类型,从而改善靶向治疗策略的设计。因此,论文所做的工作:
1. 创建泛癌症和泛组织的单细胞转录组学图谱,揭示了恶性细胞、癌前细胞以及癌症相关的基质细胞和内皮细胞中的异质性表达模式。
2. 构建名为 神农 的深度学习框架,用于针对每个细胞簇进行抗癌药物的计算机筛选。
3. 预测单个细胞对药物化合物的反应,评估药物候选分子对组织的损伤效应,并研究它们的相应作用机制。
2. 材料与方法
2.1 患者与样本收集
患者知情同意
伦理批准
样本采集与保存
- 所需的癌症及其邻近癌旁组织样本在手术后从患者处获取。
- 样本被保存在 DMEM(杜氏改良鹰培养基,ThermoFisher)中,存储温度为 4°C。
- 单细胞RNA测序(scRNA-seq)在 2小时 内完成。
临床信息
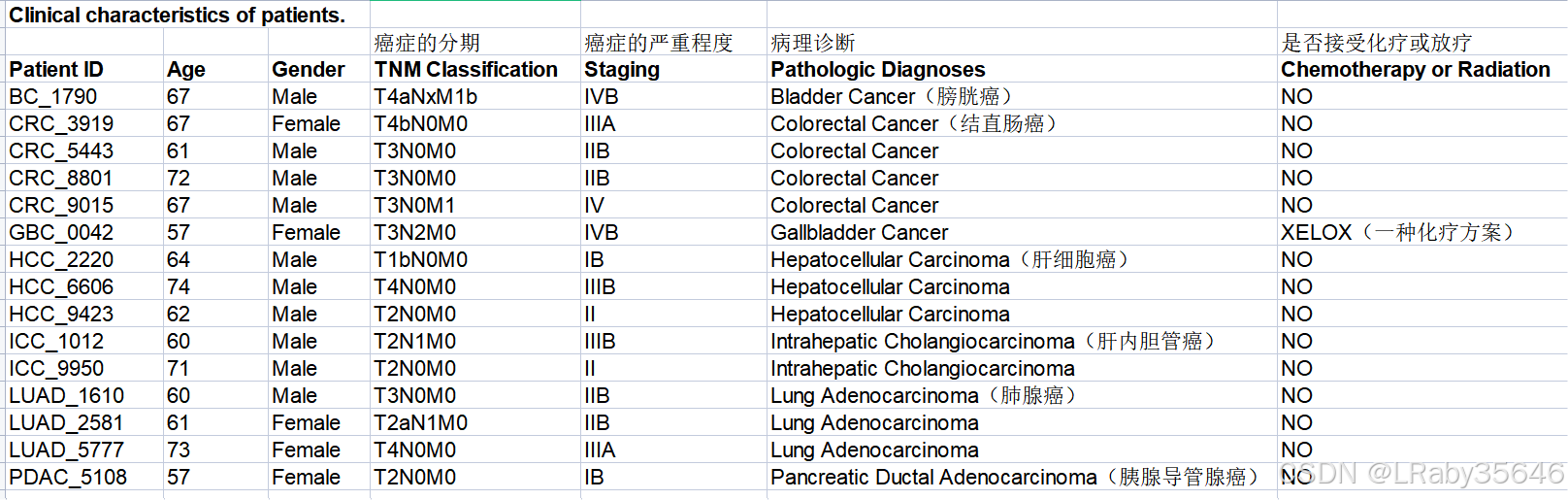
- 患者的详细临床信息列在 Table S1 中(如下图)。
2.2 单细胞测序与数据预处理
单细胞RNA数据获取
- 使用 Microwell-seq 来获得每个样本的单细胞RNA数据(这个技术好像是他们团队自己开发的)
Microwell-seq 工艺步骤
样本测序
- 样本在 MGI DNBSEQ-T7 上进行测序。
数据处理
- 原始 Microwell-seq 数据按他们之前发布的工作 [7,63] 中的协议处理。
- 测序数据对 Homo sapiens GRCh38 基因组 进行比对
数据分析
- 数据经过过滤后,使用 Seurat 和 Scanpy 对处理后的差异基因表达(DGE)数据进行了:降维分析、聚类分析、差异基因表达分析。
Tips: Microwell-seq 工艺 和 MGI DNBSEQ-T7 是两个不同的步骤,分别负责 文库构建 和 数据获取(测序)
Microwell-seq 工艺:
细胞分选:每个细胞被单独分配到微孔中,进行后续的裂解和反转录等步骤。
文库制备:通过一系列扩增和标记过程生成每个细胞的 cDNA 文库,并给每个细胞加上条形码,确保每个细胞的数据可以被唯一识别。
MGI DNBSEQ-T7 测序:
DNA 纳米球测序:在完成文库制备后,这些文库样本会被送入 MGI DNBSEQ-T7 测序仪,进行高通量的测序。
通过 DNBSEQ 技术,对文库中的每个 cDNA 分子进行读取,生成每个细胞的转录组数据。
2.3 恶性细胞鉴定
使用三种方法同时进行恶性细胞鉴定
方法一:使用 Seurat pipeline 对 DGE 数据进行聚类
每个患者的 差异基因表达(DGE)数据 被合并并使用 Seurat 管道进行聚类。分为5种细胞类型
上皮细胞(EPCAM, KRT);内皮细胞(ENG, VWF, PLVAP);基质细胞(DCN, COL1A1);淋巴细胞(CD3D, CD8A, FOXP3, MS4A1, IGKC, JCHAIN);髓系细胞(CD1C, IL1B, C1QA, S100A8, TPSAB1)
方法二:使用 inferCNV 进行推断
对每个患者的所有上皮细胞使用 inferCNV 包进行 RNA 基于拷贝数变异推断。
方法三:对潜在恶性细胞进行降维处理
-
对来自不同癌症类型的潜在恶性细胞进行 降维处理。
-
不同癌症类型的潜在恶性细胞应当形成独立的簇。
Tips:我们发现,作者只针对上皮细胞进行恶性细胞的鉴定。原因是:乳腺癌(BC)、结直肠癌(CRC)、肝细胞癌(HCC)、胆管癌(ICC)、肺腺癌(LUAD)、胰腺导管腺癌(PDAC) 的恶性细胞大多数都是来源于上皮细胞,因此它们的恶性细胞通常被归类为上皮来源的肿瘤。📌 为什么这些癌症的恶性细胞来源于上皮细胞?因为它们的起源细胞是上皮细胞,而不是来自于结缔组织或肌肉等其他组织。
但是不同的疾病恶性细胞来源不一样哦😊
2.4 神农框架
端到端模型:整个过程从输入到输出是自动化的,即输入直接通过模型传递,经过一系列操作后直接生成最终结果。
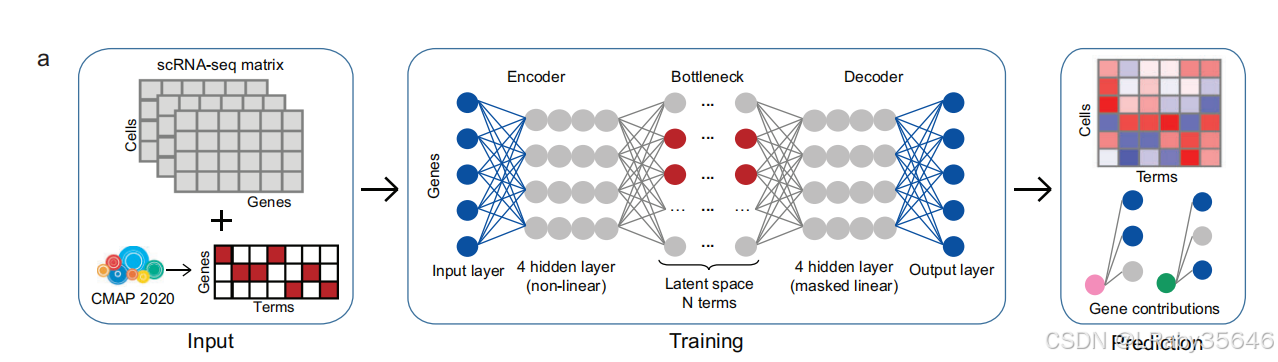
第一阶段:数据整合和扰动特征提取
-
将合并的 scRNA-seq 计数矩阵 与预处理的 扰动数据 结合,得到扰动变化特征。
-
二元矩阵表示基因表达变化:
-
使用 二元矩阵 来表示每个术语的显著差异表达特征是否在 scRNA-seq 数据中表达。
-
在该二元矩阵中,行表示术语,列表示细胞。每个元素为
1或0,表示该基因在该细胞中是否被显著表达。
-
第二阶段:建立细胞扰动预测模型
-
建立了一个细胞扰动预测模型——神农,用于捕捉细胞对药理扰动的反应,且在单细胞水平上进行分析。
-
细胞扰动预测模型建立:
-
Shennong 框架采用了 变分自编码器(VAE) 架构,捕捉细胞对药理扰动的反应。
-
模型基于 公开的 expiMap 模型,结合了来自不同条件的单细胞数据,确保能够充分捕捉术语的变异性。
-
-
编码器与解码器结构:
-
编码器网络包含 4 个隐藏层,使用 非线性激活函数,增强灵活性。
-
解码器网络按相反顺序设置相同的层,采用 被掩蔽的线性解码器,增强解码器的互操作性。
-
-
潜在空间维度与术语数量一致:
-
在瓶颈网络中,潜在空间的维度等于术语的数量,确保潜在空间能够捕捉到每个术语的特征。
-
-
训练数据:
-
模型在参考的 scRNA-seq 数据 和 扰动数据 上进行训练。
-
细胞来源标签(如肿瘤、邻近组织或正常组织)通过额外的向量对每个细胞进行编码。
-
扰动数据的二元矩阵由表示术语的一组潜在变量编码。
-
-
条件与注意力机制:
-
由于扰动数据的规模较大且可能存在冗余信息,模型在潜在空间中实现了类似 注意力机制,通过 群体套索正则化 来选择每个细胞的扰动影响术语。
-
-
解码器训练:
-
经过 修剪和丰富 后,术语被输入到 线性解码器 中。解码器网络通过术语矩阵来设计神经网络架构,并将模型参数与术语特定的潜在维度进行连接。
-
Encoder:
Input -> [512] -> [512] -> [512] -> [512] -> [2000]
Decoder:
Input (Latent Space) -> [2000] -> [8500]
(plus 2D condition input)
第三阶段:影响测量与基因贡献评估
-
扰动影响的度量:
-
训练后的端到端模型用于度量每个术语对每个细胞的影响。
-
在预测过程中,训练集和预测集的潜在表示被映射到同一潜在空间,并修正其中的 批量效应(batch effects)。
-
-
术语的影响评分:
-
每个术语的影响评分通过 潜在空间中的潜在分数 和 方向 来度量,结果存储在矩阵中。
-
差异术语通过 Bayes 检验 在细胞簇的术语层面进行识别。
-
-
统计显著性与富集评分:
-
如果某个术语的 绝对对数 Bayes 得分 大于或等于 2.3,则该术语在某个细胞类型中被认为是显著差异的。
-
这种得分称为 富集评分,表示术语在细胞簇中的影响。
-
-
基因对术语的贡献:
-
提取并排序每个术语的解码器权重的 绝对值,以度量每个术语的基因贡献。
-
权重的绝对值越高,表示该基因受术语的影响越大,反映了基因在术语中的相对重要性
-
3. 数据获取
-
原始序列数据存储:
-
本文报告的原始序列数据已存储在 中国国家基因组数据中心(China National Center for Bioinformation)/ 北京基因组研究所(Beijing Institute of Genomics),中国科学院基因组序列档案库(Genome Sequence Archive,GSA)中。
-
数据的存储编号为 GSA-Human: HRA006591,可以通过 https://ngdc.cncb.ac.cn/gsa-human 公开访问。
-
-
数据合规性:
-
存储并公开的数据符合 中国科技部 的相关规定。
-
-
处理后的计数矩阵和细胞注释:
-
处理后的 计数矩阵 和 细胞注释数据 可在 figshare 上获取,链接:https://figshare.com/s/ac34f719115943d1d46c。
-
-
LUAD 和 PDAC 数据来源:
-
LUAD 和 PDAC 的单个患者 scRNA-seq 数据 来自 基因表达综合数据库(Gene Expression Omnibus,GEO)。
-
LUAD 数据的登录号为 GSE131907,PDAC 数据的登录号为 GSE155698。
-
-
细胞类型标签和元数据:
-
所有 细胞类型标签 和 元数据 来自 原始公开文献。
-
-
第三方数据对比:
-
对于第三方对比的 scRNA-seq 数据,LUAD 数据来自 ArrayExpress 数据库,登录号为 E-MTAB-6149。
-
HCC 数据来自 GEO,登录号为 GSE1496140。
-
-
细胞类型标签的重新聚类和注释:
-
元数据来自 原始文献,细胞类型标签根据这些文献进行了 重新聚类 和 注释。
-
4. 代码获取
-
源代码:
-
用于重现分析和训练 Shennong 框架 的 源代码 已公开。
-
-
代码存储位置:
-
代码可以在以下平台访问:
-
figshare:https://figshare.com/s/ac34f719115943d1d46c(这个比Github中的数据更全,如果想复现代码,建议从这里面下载)
-
-
5. 结果解读
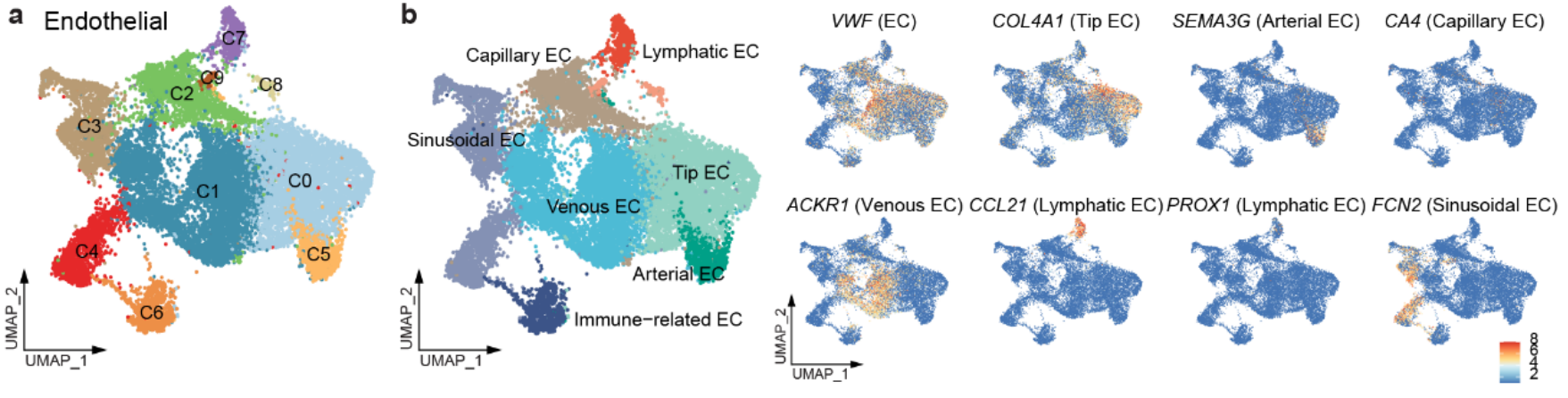
5.1 使用Microwell-seq构建泛癌单细胞图谱
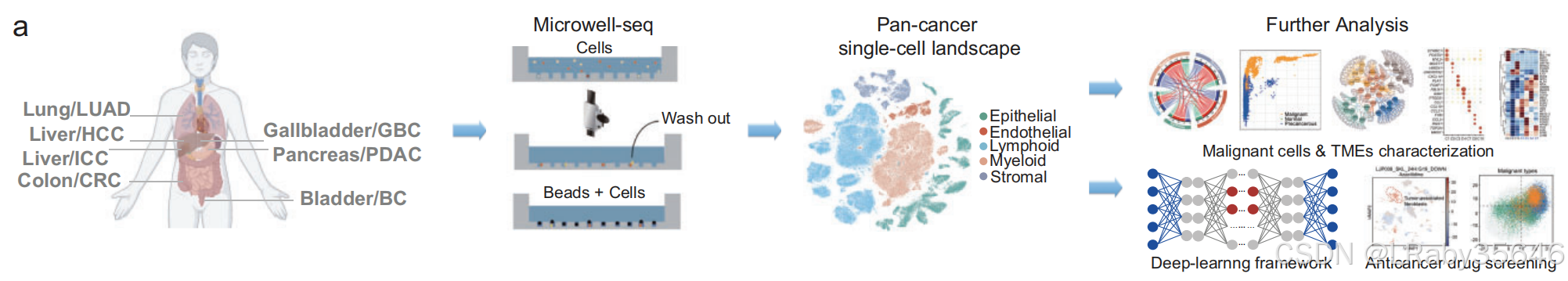
数据收集与处理
-
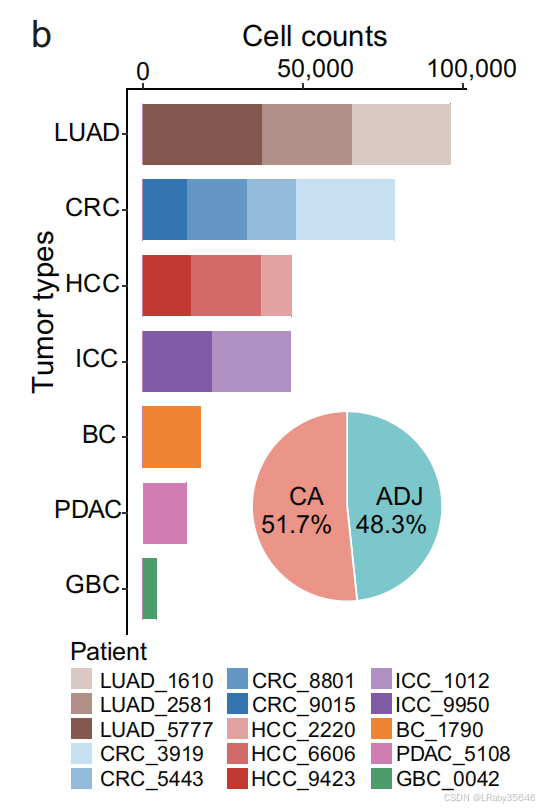
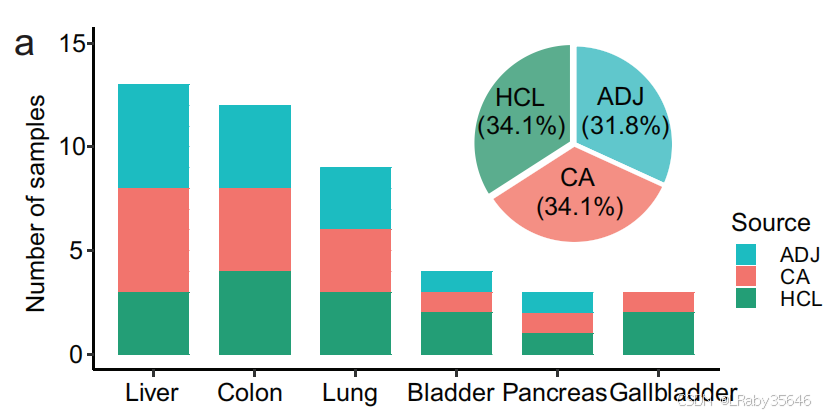
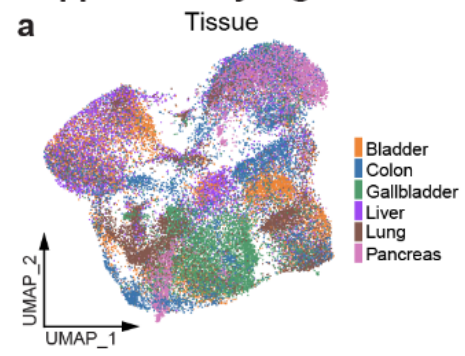
从 15 位患者 收集了来自 7 种肿瘤类型 的细胞数据,共计 303,351 个细胞。
-
其中,51.7% 的细胞 来源于 肿瘤组织。48.3%的细胞来源于癌旁
-
肿瘤类型:肺腺癌(LUAD)、结直肠癌(CRC)、肝细胞癌(HCC)、肝内胆管癌(ICC)、膀胱癌(BC)、胰腺导管腺癌(PDAC) 和 胆囊癌(GBC)
无监督聚类与细胞集群
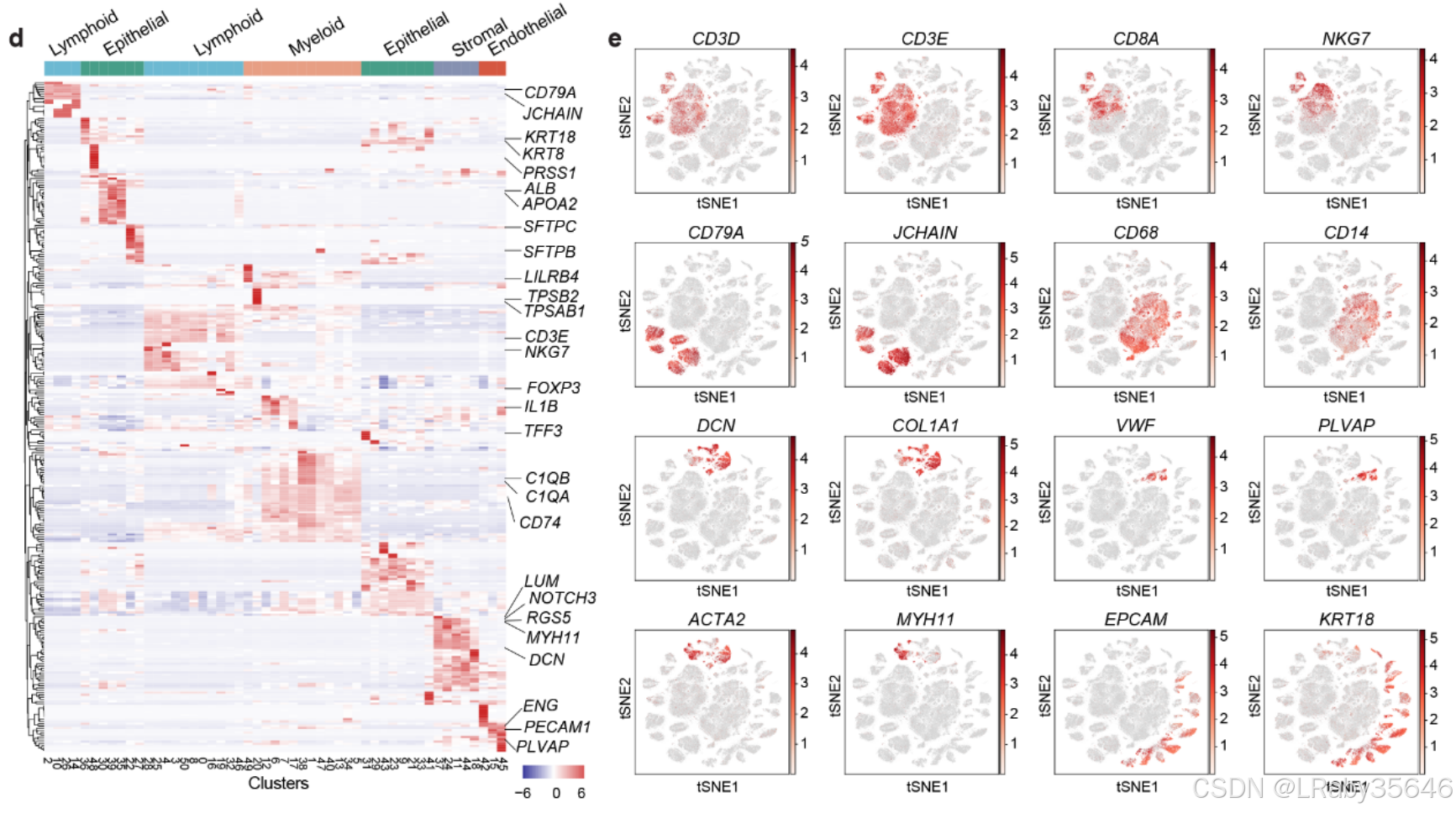
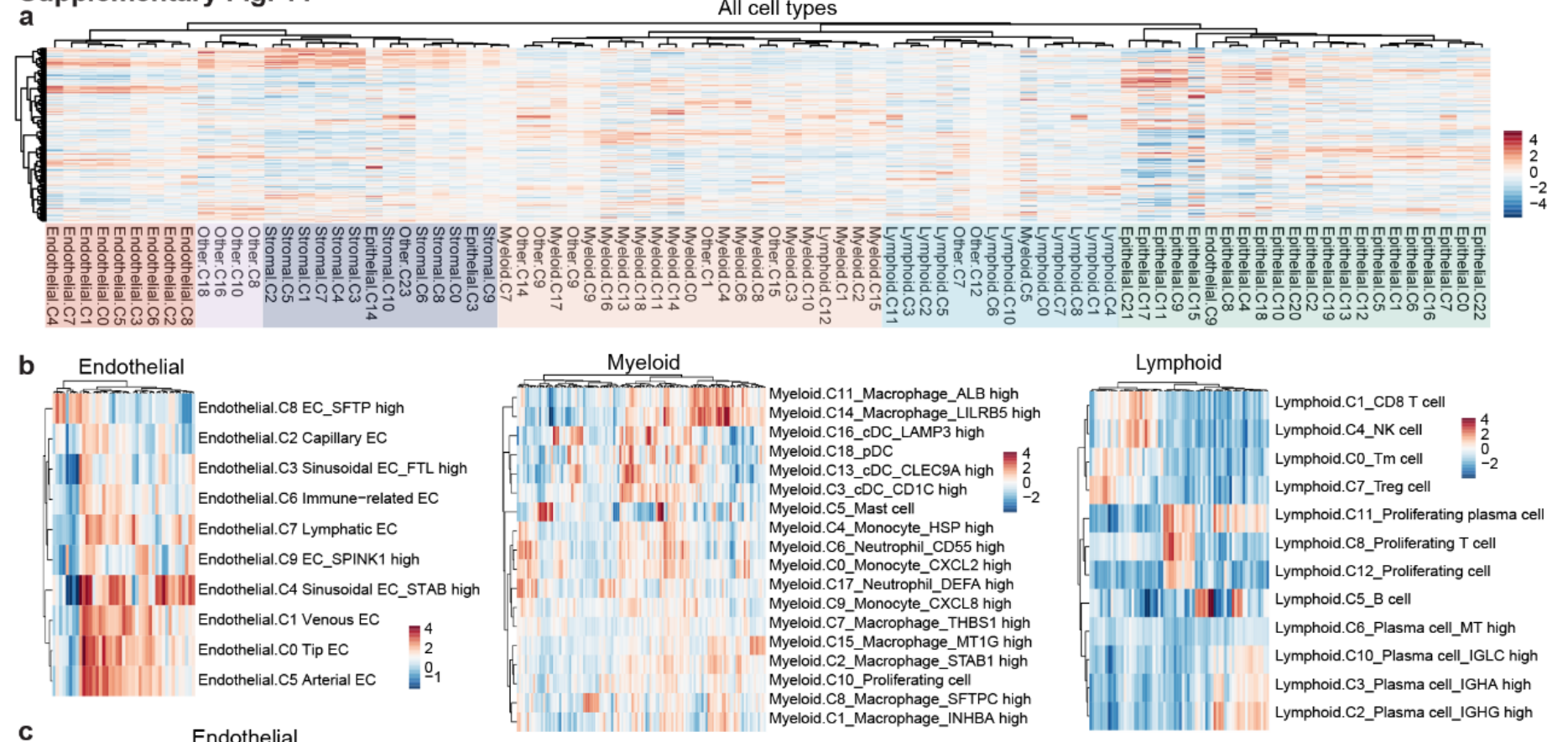
- 使用 无监督聚类,识别了 51 个主要细胞集群(Fig1c,Fig S1a)。
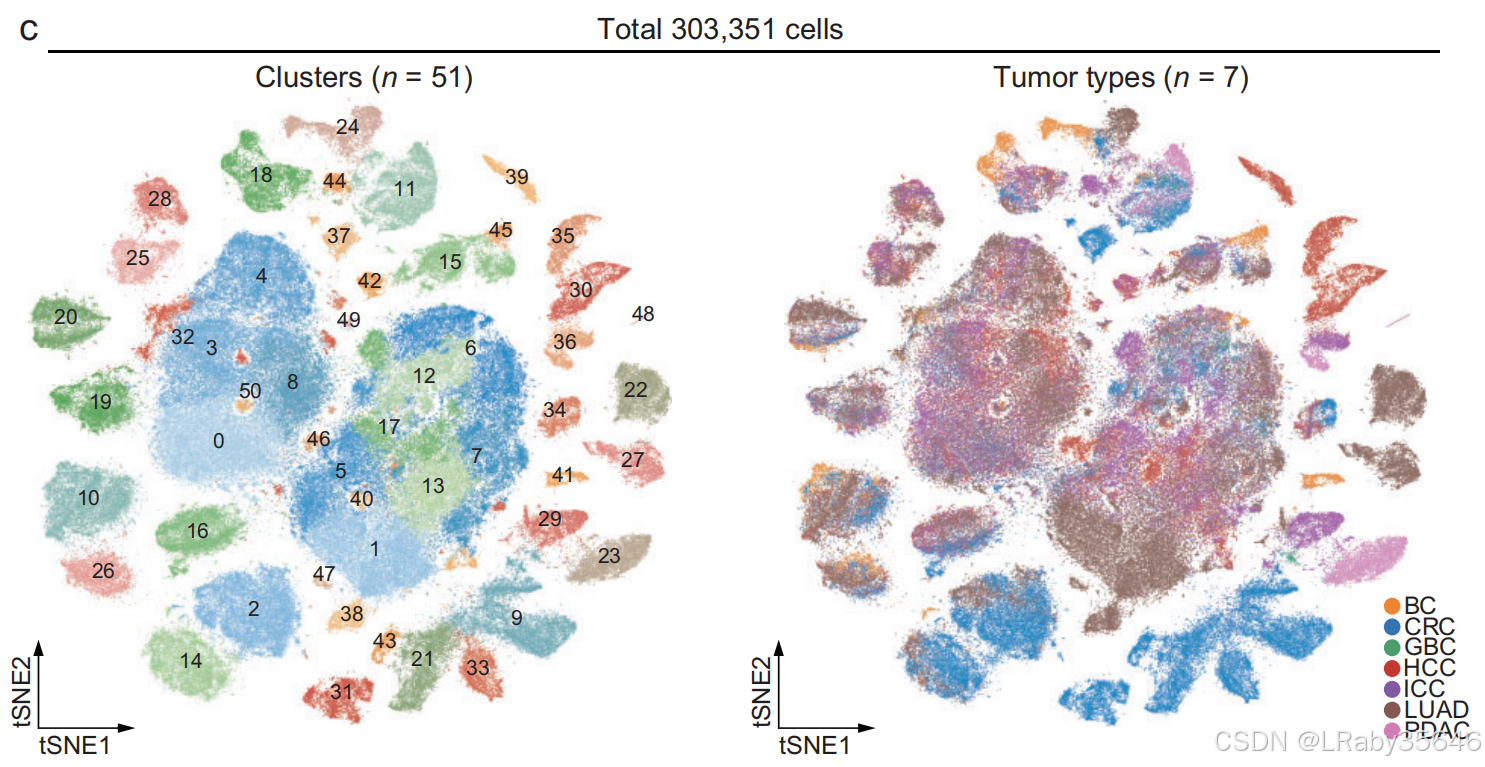
-
细胞类型分类:
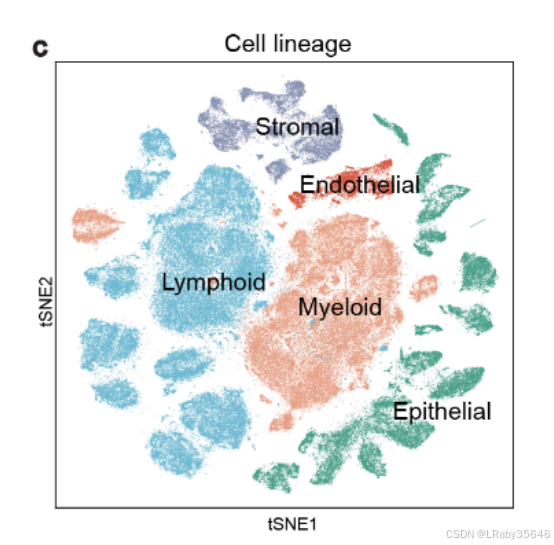
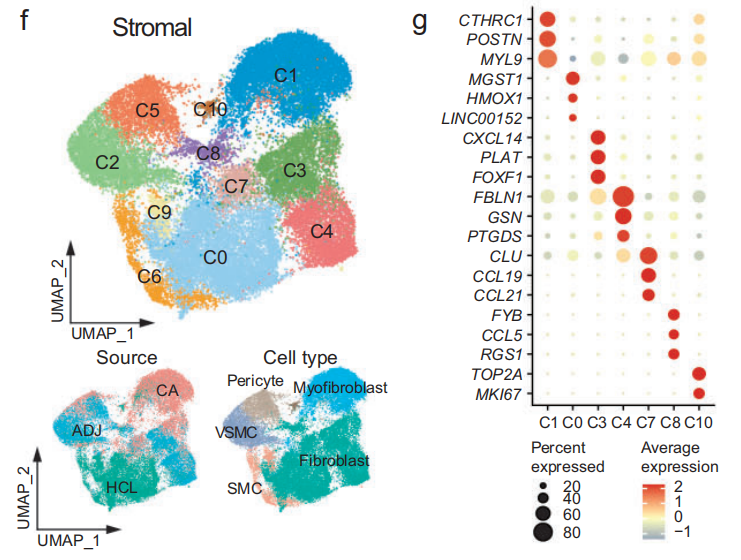
基于细胞类型特异性标记物,这些集群被划分为 5 个主要谱系(Fig S1c):-
淋巴细胞(Lymphoid):CD3D、CD3E、CD79A、JCHAIN
-
髓系细胞(Myeloid):CD68、CD14
-
基质细胞(Stromal):DCN、COL1A1、ACTA2
-
内皮细胞(Endothelial):VWF、PLVAP
-
上皮细胞(Epithelial):EPCAM、KRT18
-
另外将每个细胞类型的marker基因画了热图与t-sne图(如下图所示)
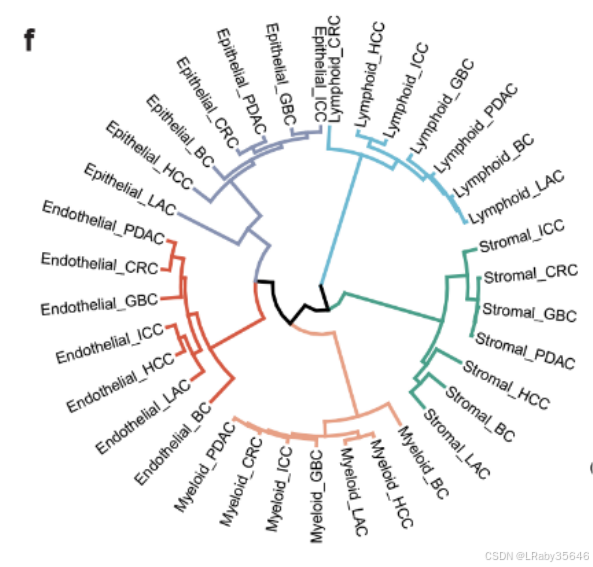
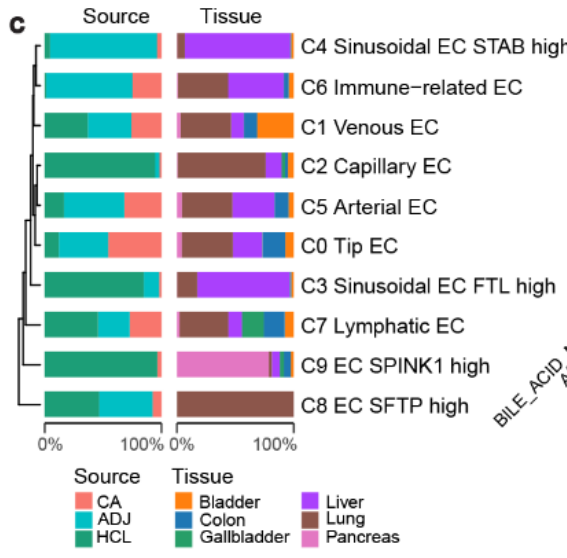
- 细胞谱系树:细胞谱系树显示,来自同一细胞谱系的细胞集群往往会在不同组织类型之间趋向于聚集(Fig 1d,Fig S1f)。[这个图我不太了解意义是什么]
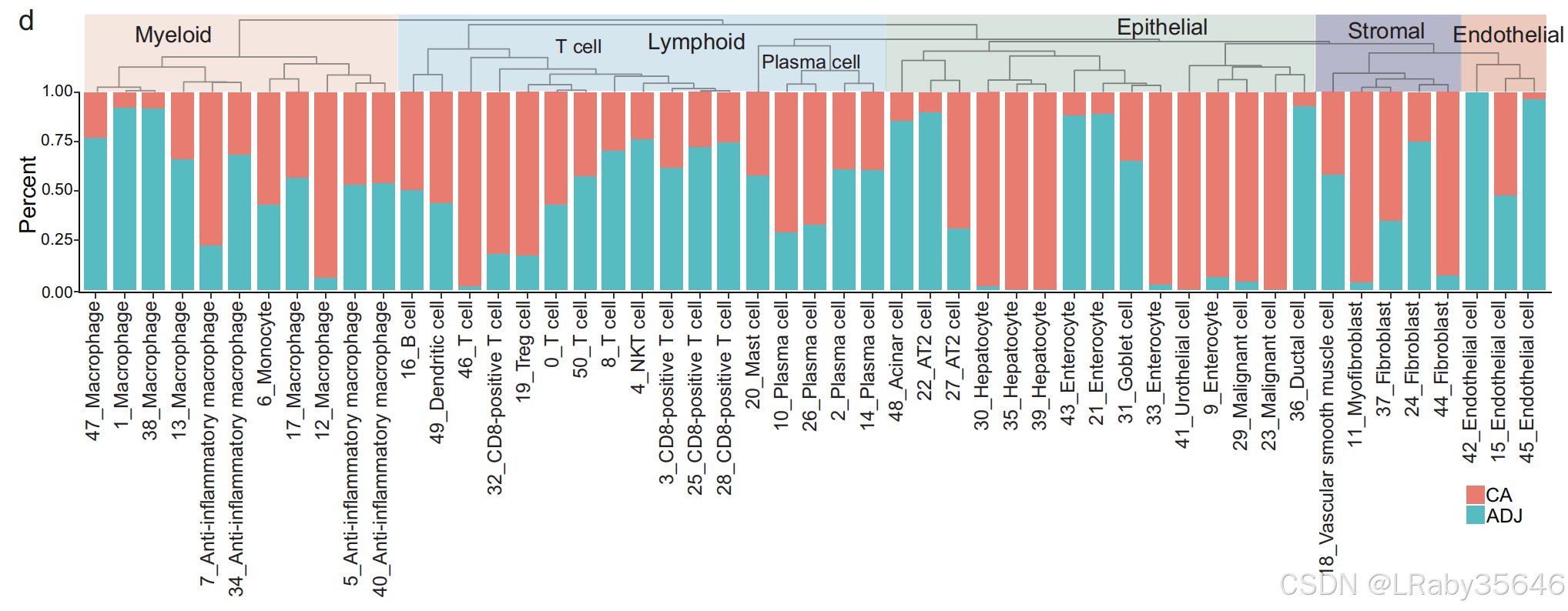
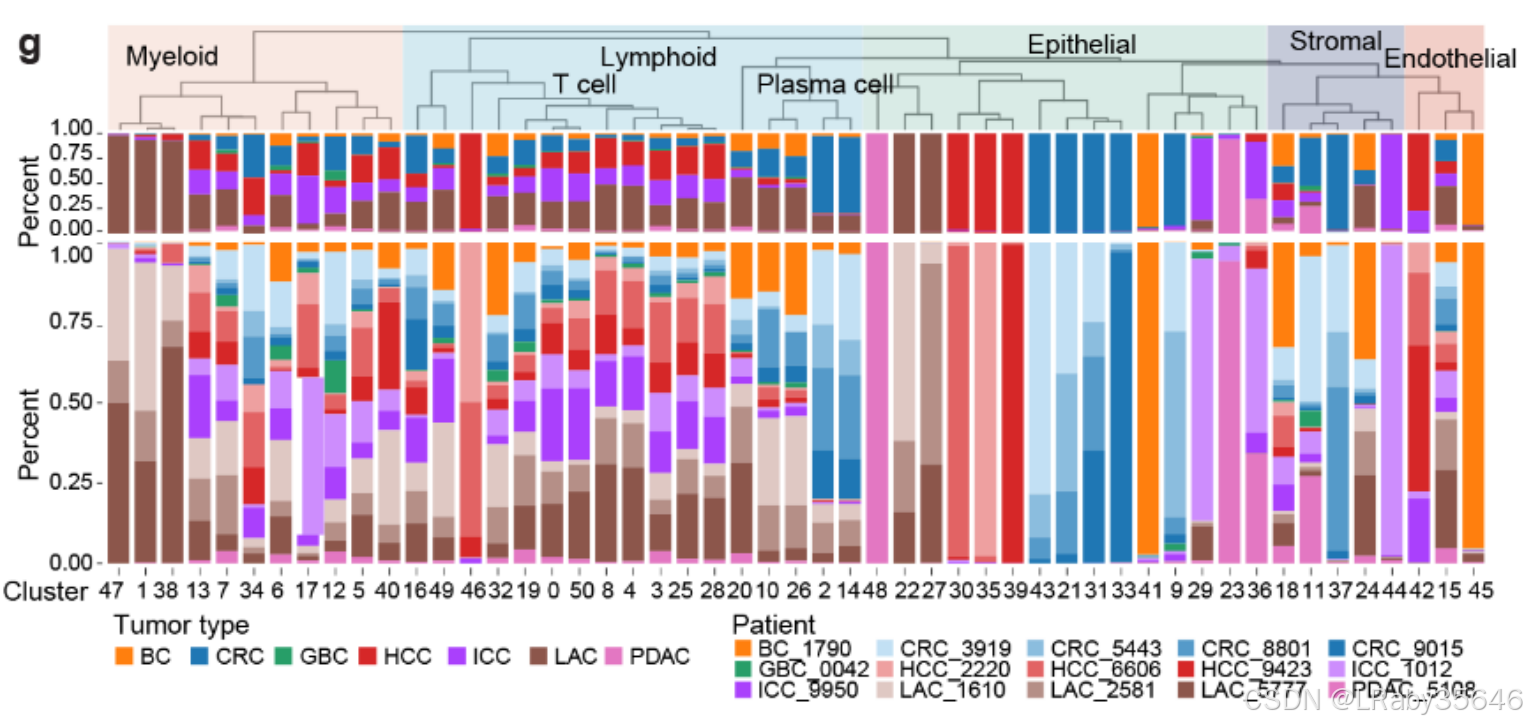
细胞集群的组成与患者特异性(Fig S1g)
上皮细胞特征:
-
几乎所有上皮细胞展示了组织类型特异性特征,并且来自多位患者。
-
某些集群展示了患者特异性模式。例如:
-
C9、C21 和 C43 主要是多位患者贡献的肠上皮细胞。
-
C33 主要由患者 CRC_9015 提供。
-
肝细胞特征:
- 每个肝细胞集群(如 C30、C35 和 C39)主要来自单一患者。
- C36 被识别为导管细胞,来自多种肿瘤类型(HCC、ICC 和 PDAC)患者。
免疫、基质与内皮细胞集群(Fig S1g)
-
基质、内皮和免疫集群:
-
来自不同肿瘤类型和患者的 基质、内皮和免疫集群 分别被很好地整合。
-
-
特定细胞集群:
-
C11 和 C24 是 肌成纤维细胞/成纤维细胞。
-
C15 和 C42 是 内皮细胞。
-
淋巴谱系主要包含 T 细胞、B 细胞 和 浆细胞。
-
髓系谱系主要包括 巨噬细胞、单核细胞 和 树突状细胞。
-
-
T 细胞分布:
-
C46 主要包含来自多位 HCC 患者的 T 细胞。
-
C1、C38 和 C47 主要来自 LUAD 患者,尤其是 C47。
-
5.2 恶性细胞和癌前细胞内的异质性表达模式
理解 肿瘤内异质性 对揭示癌症进展和提高治疗效果非常重要。这部分研究通过整合 正常组织的 scRNA-seq 数据(郭国骥老师团队之前做的工作)来研究肿瘤细胞及其 肿瘤微环境(TME) 的细胞异质性。
数据来源和技术
-
scRNA-seq 数据平台:
-
所有 scRNA-seq 数据都使用相同的平台 Microwell-seq 生成,排除了可能的 技术偏差。
-
-
数据合并:
-
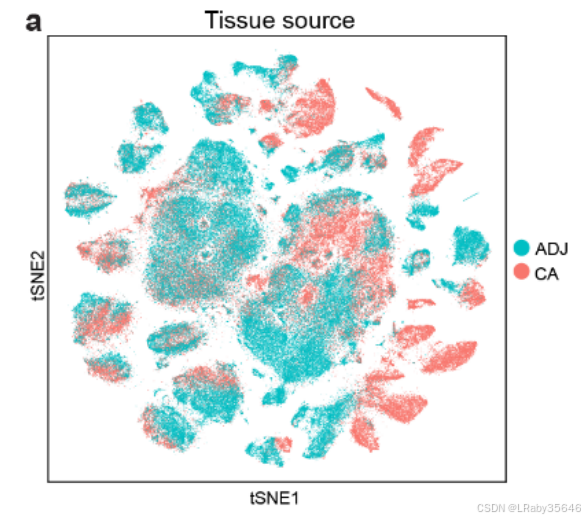
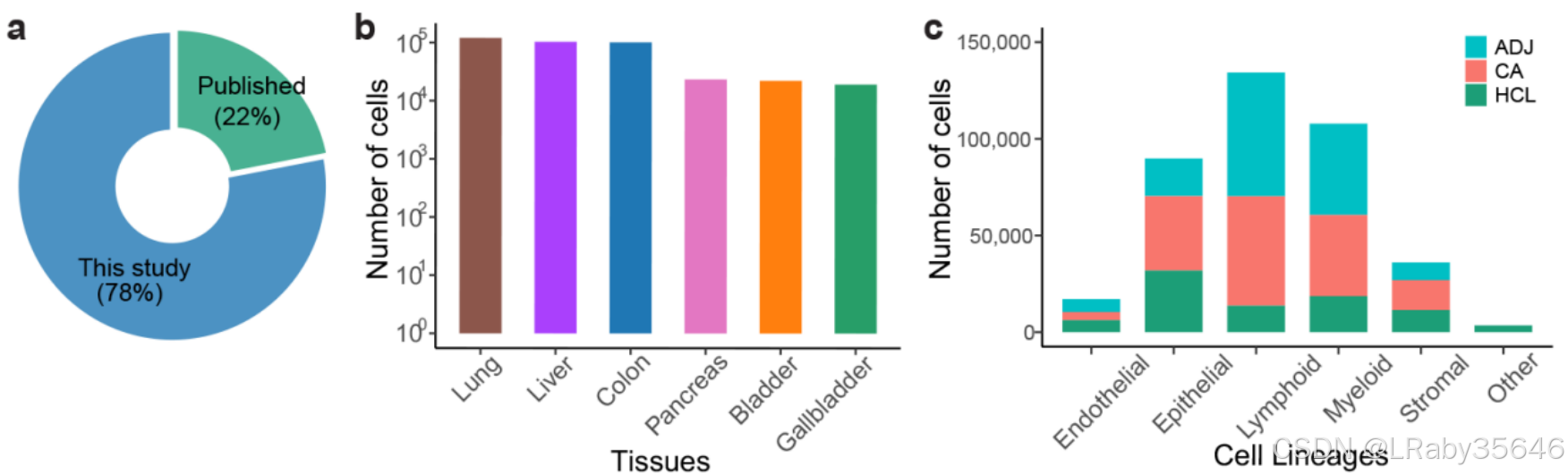
合并后的数据集包含 388,646 个细胞,其中大约三分之一来自 肿瘤组织(CA)、邻近组织(ADJ) 和 正常组织样本(Fig2a,)。
-
来自 HCL 的细胞占 22%。
-
细胞间通讯分析
-
细胞–细胞通讯:
-
细胞–细胞通讯分析显示,在 正常组织、邻近组织(ADJ) 和 肿瘤组织(CA) 中,通讯频率呈 显著增加(p < 0.05) 的趋势(Fig 2b 和Fig S2d)。
-
-
邻近组织的过渡状态:
-
这一结果表明,邻近组织可能处于 正常与肿瘤组织之间的过渡状态,可能为肿瘤的 早期发展 提供了线索。
-

随着从正常组织到肿瘤组织的过渡,细胞间的 相互作用显著增加。这表明,邻近组织和肿瘤组织中的细胞之间有 更多的交互,特别是在 基质细胞 和 免疫细胞(如髓系和淋巴细胞) 等细胞类型之间的互动。
比较 HCL 和 ADJ 中细胞类型的交互,可以看出 邻近组织(ADJ) 的细胞间互动比正常组织(HCL)有所增加。这可能意味着 邻近组织 正在经历 转变,细胞间的互动开始 接近肿瘤组织的状态。这为肿瘤的早期诊断提供了可能的线索。
肿瘤组织中的细胞间互动 强度明显增加,尤其是 基质细胞和髓系细胞、淋巴细胞 与其他细胞的互动。这反映了肿瘤微环境的 复杂性,肿瘤细胞和其他非肿瘤细胞(如免疫细胞、基质细胞)的相互作用可能为肿瘤的发展提供支持。
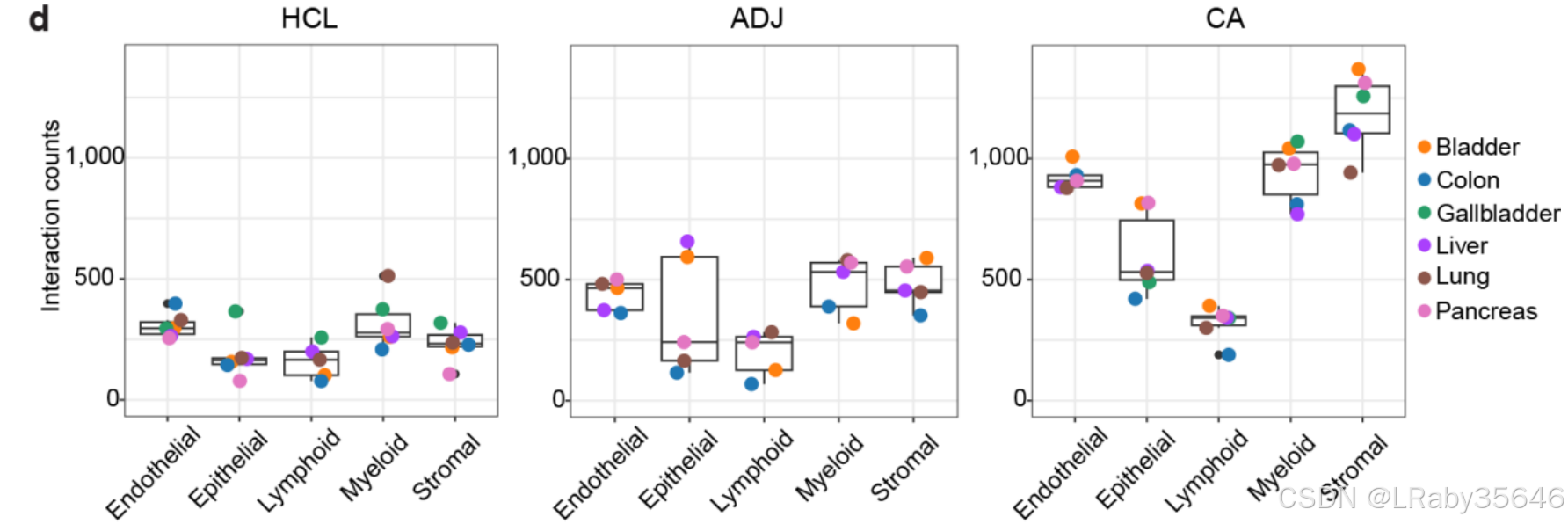
x轴:表示不同的细胞类型,y轴:表示这些细胞类型之间的 细胞–细胞交互数量。
在不同组织类型(HCL、ADJ、CA)中,细胞之间的 交互频率 有显著差异。尤其是在肿瘤组织(CA)中,细胞之间的互动更为 频繁。
肿瘤组织 中细胞的 交互作用更为频繁,尤其是 基质细胞 和 免疫细胞。
三种方法联合用于识别恶性细胞
准确的恶性细胞定义对于肿瘤异质性模式的表征至关重要。仅将细胞归类为 上皮细胞 并不足以识别肿瘤组织中的恶性细胞。
方法在上面。
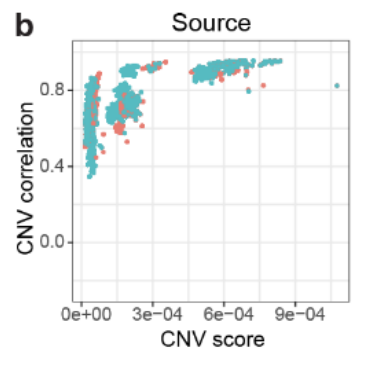
对每一个病人都进行恶性细胞的判断(Fig S3)
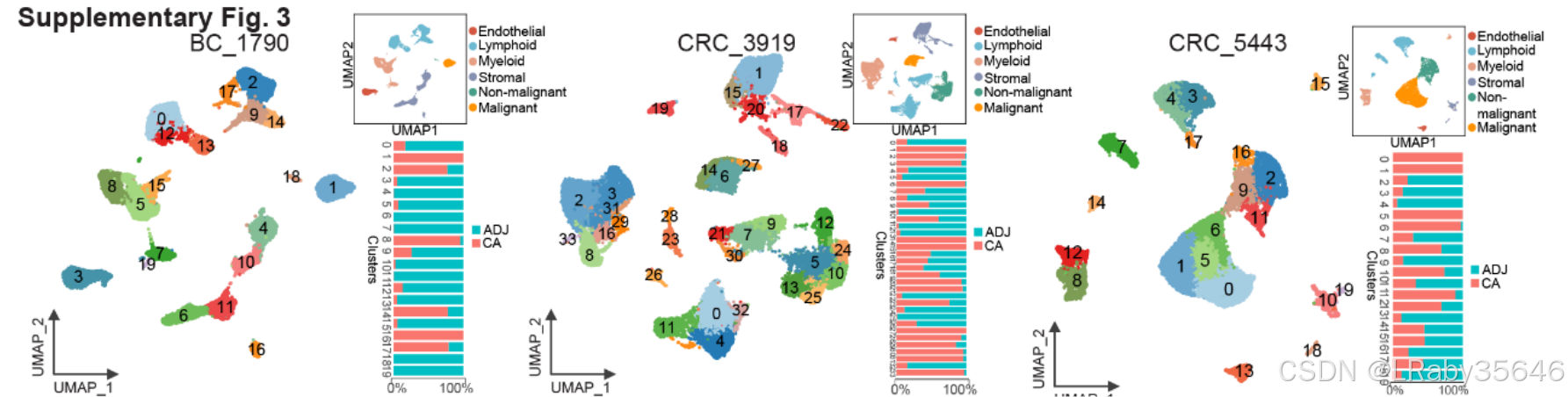
使用inferCNV将每个病人的细胞分为正常、恶性、癌前细胞(Fig S4)
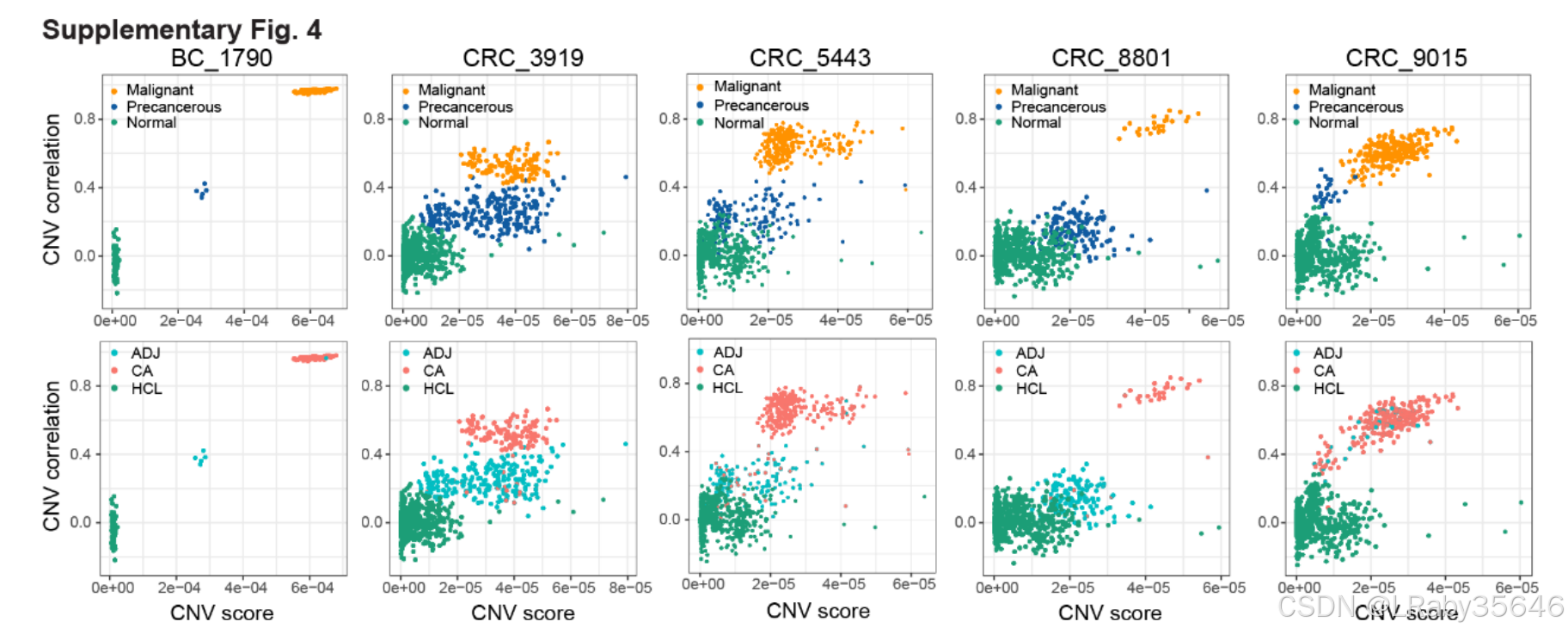
以患者 ICC_1012 为例,分析步骤如下:
-
细胞类型分配:首先将细胞分配到不同的细胞类型,并识别 不同上皮细胞集群中的高表达基因(Fig 2d)。
-
拷贝数变异(CNV)推断:然后,使用 HCL 中的正常结肠上皮细胞 作为参考,推断这些上皮细胞集群中的 CNV 分数(Fig 2e)。
-
维度降维分析:最后,利用维度降维方法,识别出 潜在的恶性细胞集群,并发现它们与非恶性上皮细胞分开聚类。
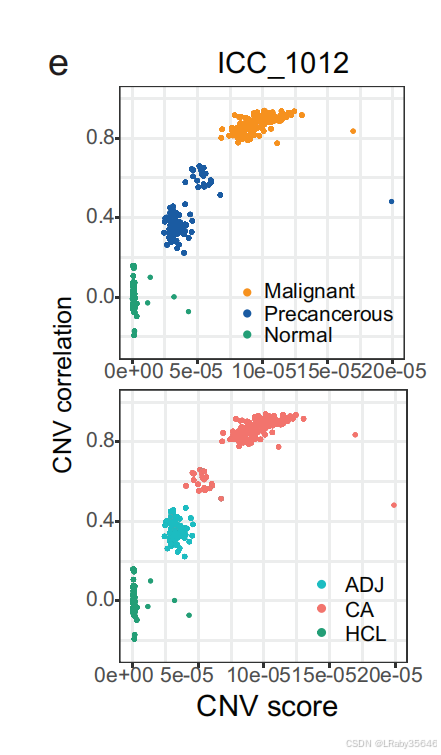
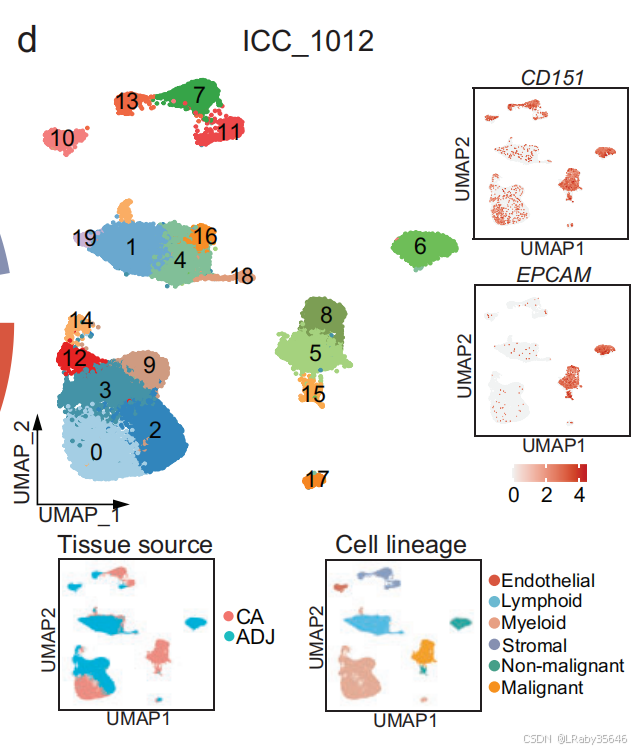
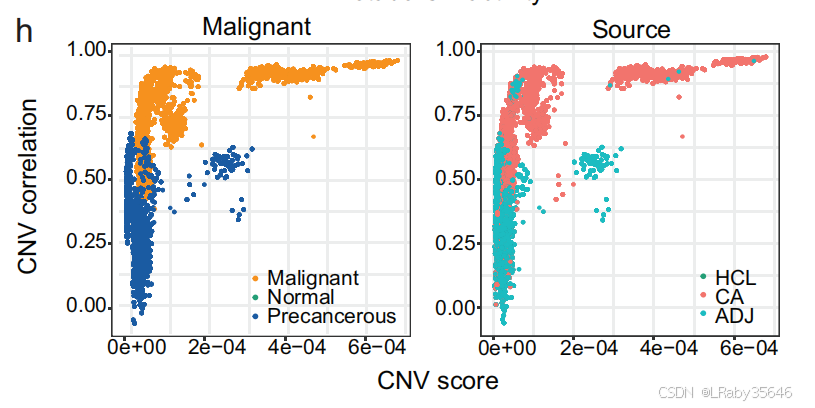
与非恶性上皮细胞相比,恶性细胞表现出 更高的 CNV 分数,并在 ICC_1012 中形成 独立的集群。尽管这些上皮细胞中的 EPCAM 和 CD151(上皮细胞标志基因)被过度表达,但恶性细胞的特征依然明显不同。这里并不知道癌前细胞是如何识别出来的,下面马上介绍。
癌前细胞的识别
几乎所有的 非恶性上皮细胞 都表现出 癌症相关基因的高表达,并且具有 边缘的 CNV 分数,这表明这些细胞可能处于肿瘤进展的早期阶段。
例如,在以下集群中观察到这种现象:
BC_1790 的 C18 集群
ICC_1012 的 C6 和 C15 集群
PDAC_5108 的 C4、C11 和 C20 集群
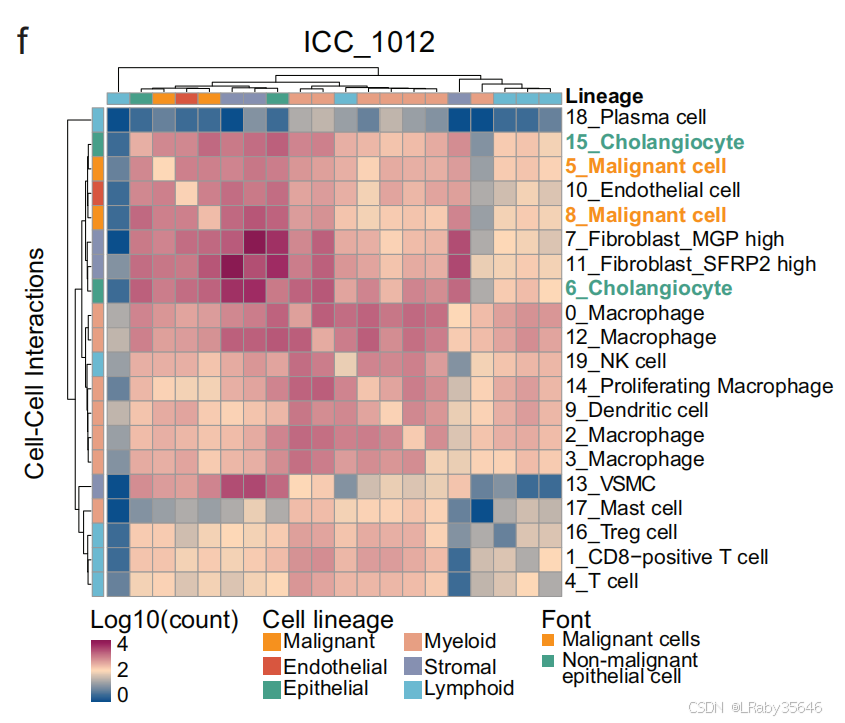
1. 细胞间通讯和代谢活动分析(以 ICC_1012 为例)
-
细胞间通讯(Fig 2f) 和 代谢活性分析(Fig 2g) 表明,这些 非恶性上皮细胞 显示出与 恶性细胞相似的特征:
-
它们与 肿瘤微环境(TME) 的 相互作用较强。
-
氧化磷酸化 下调,同时 糖酵解和糖异生 上调。
-
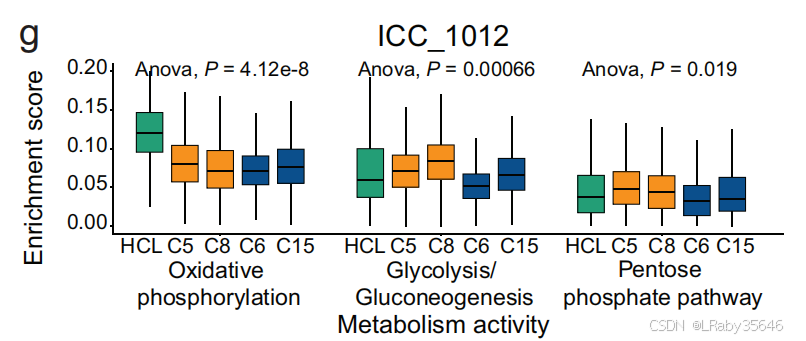
2. 细胞来源与聚类分析
-
大多数这些细胞来源于 邻近组织,并未与恶性细胞聚集在一起。
-
非恶性上皮细胞 来自 肿瘤组织 的表现出 中间的 CNV 分数,例如:
-
ICC_1012 中的 C15
-
LUAD_1610 中的 C13 和 C22
-
LUAD_5777 中的 C16 和 C20
-
PDAC_5108 中的 C20(Fig 2d、2e 和Fig S3、S4)
-
3. 上皮-间质转化(EMT)
-
PDAC_5108 中的 C20 集群 可能正在经历 上皮-间质转化(EMT),这一过程通过 EMT 分数 最高的表现得到了确认(Fig S5d)
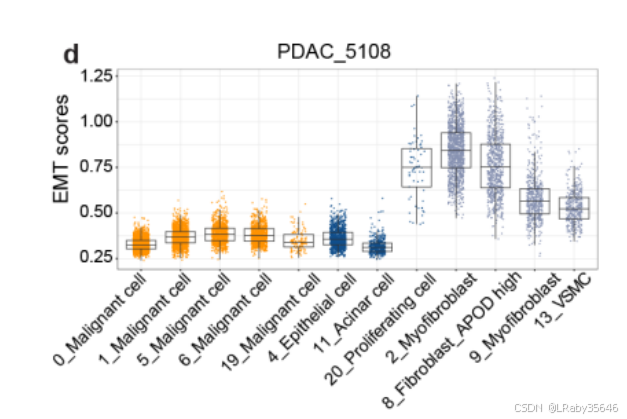
4. 数据验证
这些观察结果得到了已发布的 scRNA-seq 肿瘤数据集 的支持,这些数据集来自 10xChromium 平台(Fig S5e–j),并使用了 Tabula Sapiens 中对应的健康正常组织作为对照。(为什么要跟Tabula Sapiens 中对应的健康正常组织对比,他们不是自己有上皮细胞的正常组织吗?Fig 2a和Fig S2a-c)
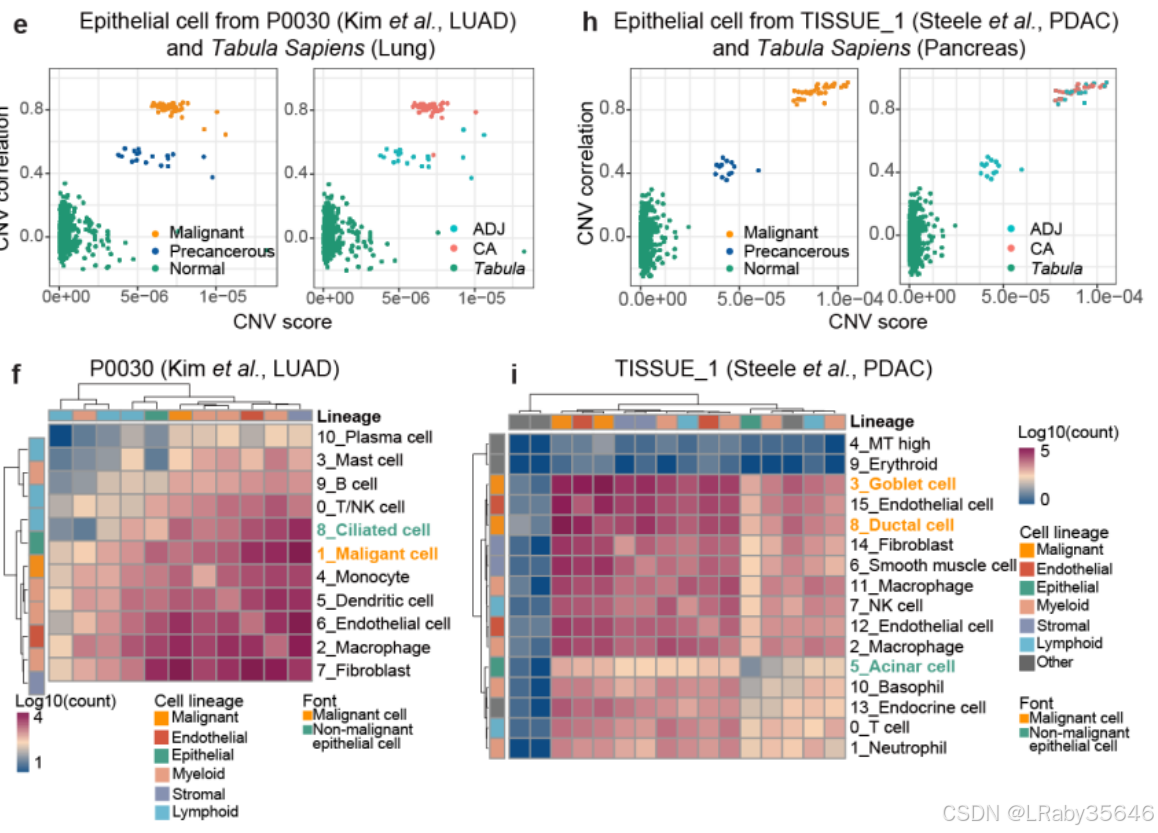
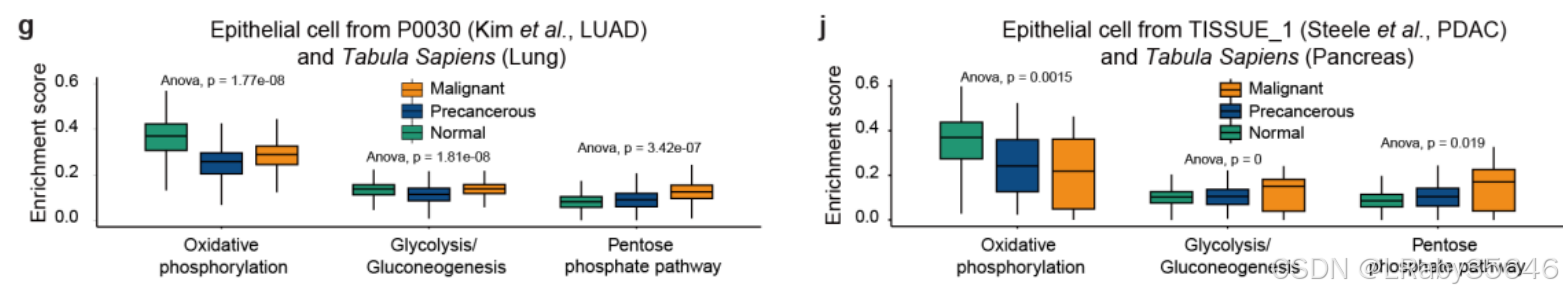
通过 CNV 分析、肿瘤标志基因表达、细胞间通讯和代谢活性评估,推测 非恶性上皮细胞 可能是 癌变的早期阶段,表示 从正常组织到肿瘤组织的过渡状态。将这些细胞称为 “癌前细胞”,并与正常上皮细胞和恶性细胞区分开。识别了 34,926 个高可信度的恶性细胞 和 23,034 个非恶性(癌前)细胞,这些细胞分布在 全癌症景观 中(Fig 2h)
恶性、癌前和正常细胞的聚类与调控模式分析
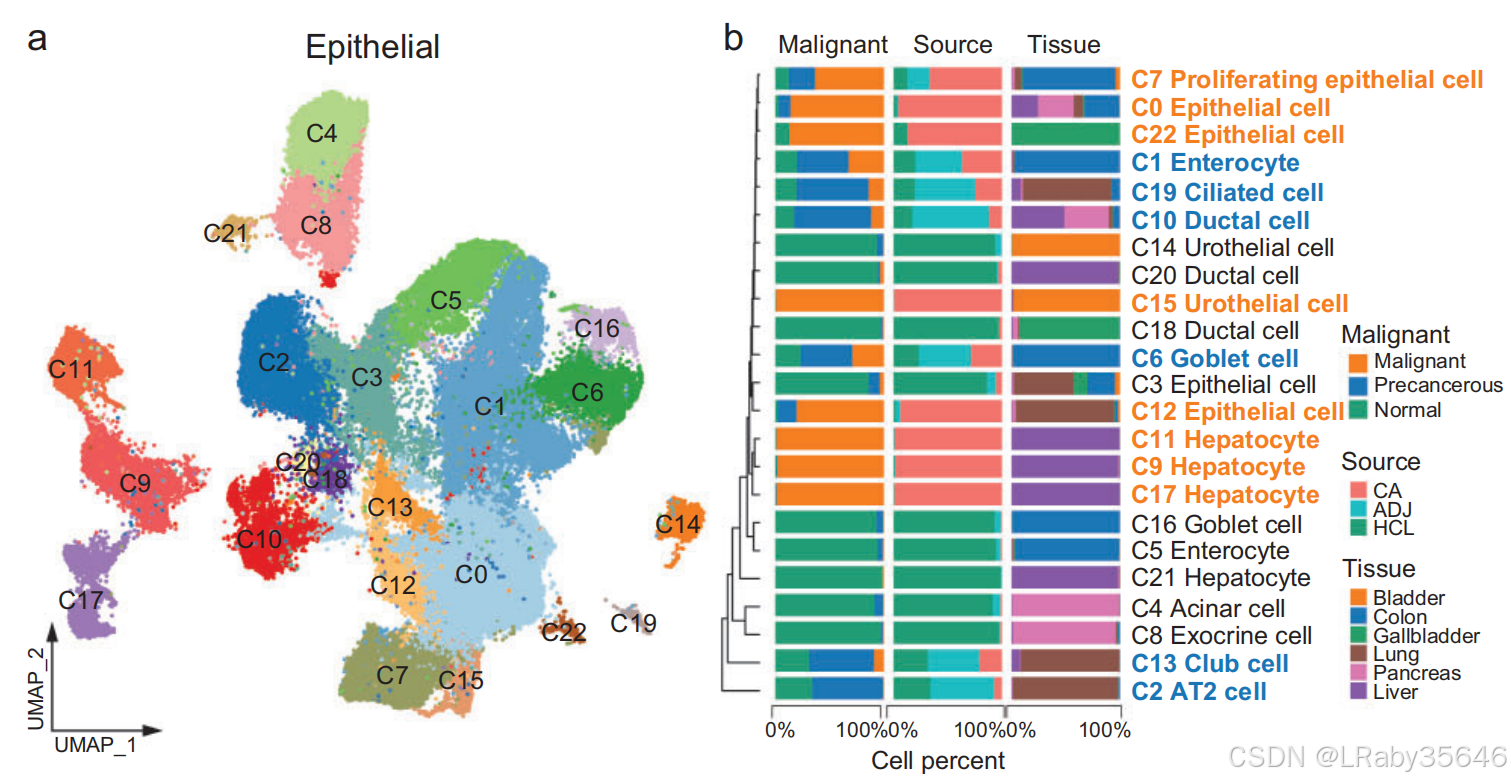
重新聚类了来自每个患者的 恶性、癌前(癌变前) 和 正常上皮细胞,并将其整合到 全癌症景观 和 HCL 数据中。
恶性细胞、癌前细胞 和 正常上皮细胞 在总细胞中的比例分别为 38.9%、25.6% 和 35.5%。
1. 聚类分析与细胞标注
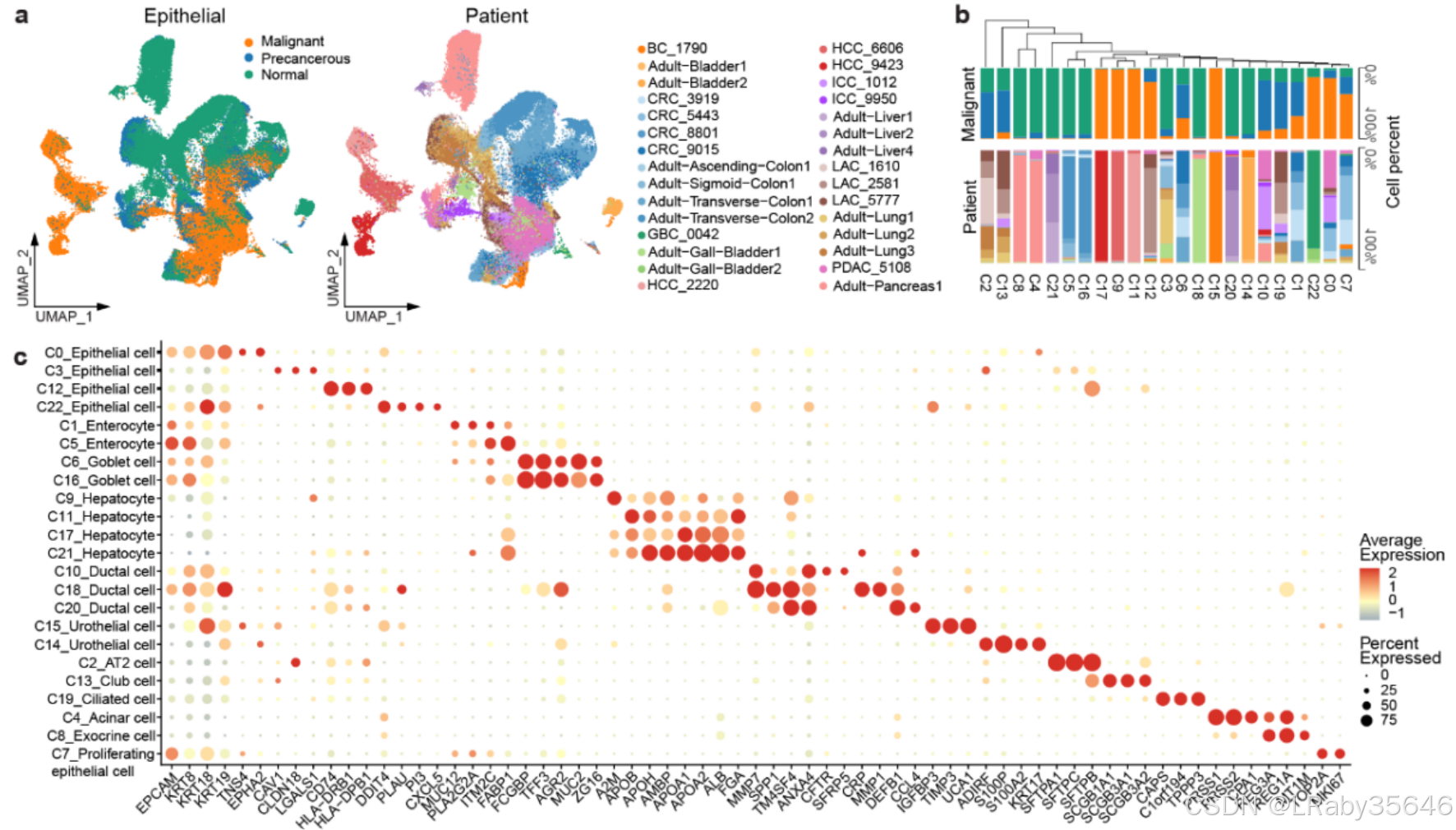
通过 无监督聚类,我们识别出 23 个集群(C0–C22),并根据 细胞特异性标记基因 对其进行了注释(Fig 3a、3b 和Fig S6a–c)
-
正常细胞占主导的集群(如 C3、C5、C20、C21、C16)包含来自多个 供体和组织 的细胞,表明 批次效应最小。
-
恶性细胞占主导的集群(如 C9、C11、C17、C12、C15、C22)主要由 单一患者 的细胞组成,显示出 广泛的肿瘤间异质性。
2. 癌前细胞的聚类与特征
-
癌前细胞 聚集在正常细胞或恶性细胞附近,形成来自特定组织(如 C2、C6、C10、C19)或结构(如 C13)的 独立集群,反映了肿瘤进程中的 不同过渡状态(Fig3a-b)。
3. 代谢和信号通路分析
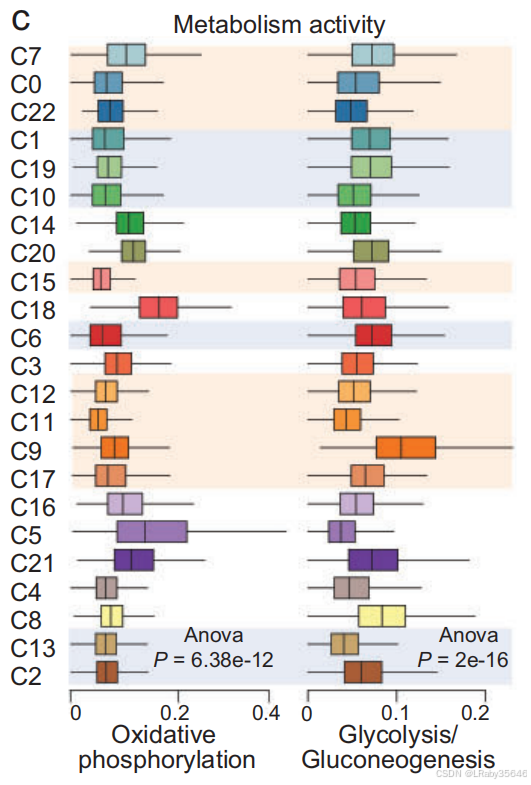
-
代谢分析 和 肿瘤标志物 研究显示,恶性集群 和 癌前集群 中的 氧化磷酸化 被下调,而 糖酵解 和 mTORC1 信号通路 被上调,这与 癌细胞的生物能学特征 相一致。
-
细胞增殖相关基因集 在恶性细胞集群中富集,包括 E2F 目标基因、G2M 检查点 和 MYC 目标基因。
-
与肿瘤发育和进展相关的通路,如 Wnt、Notch 和 TGF-β,在 恶性 和 癌前细胞集群 中也有所富集。
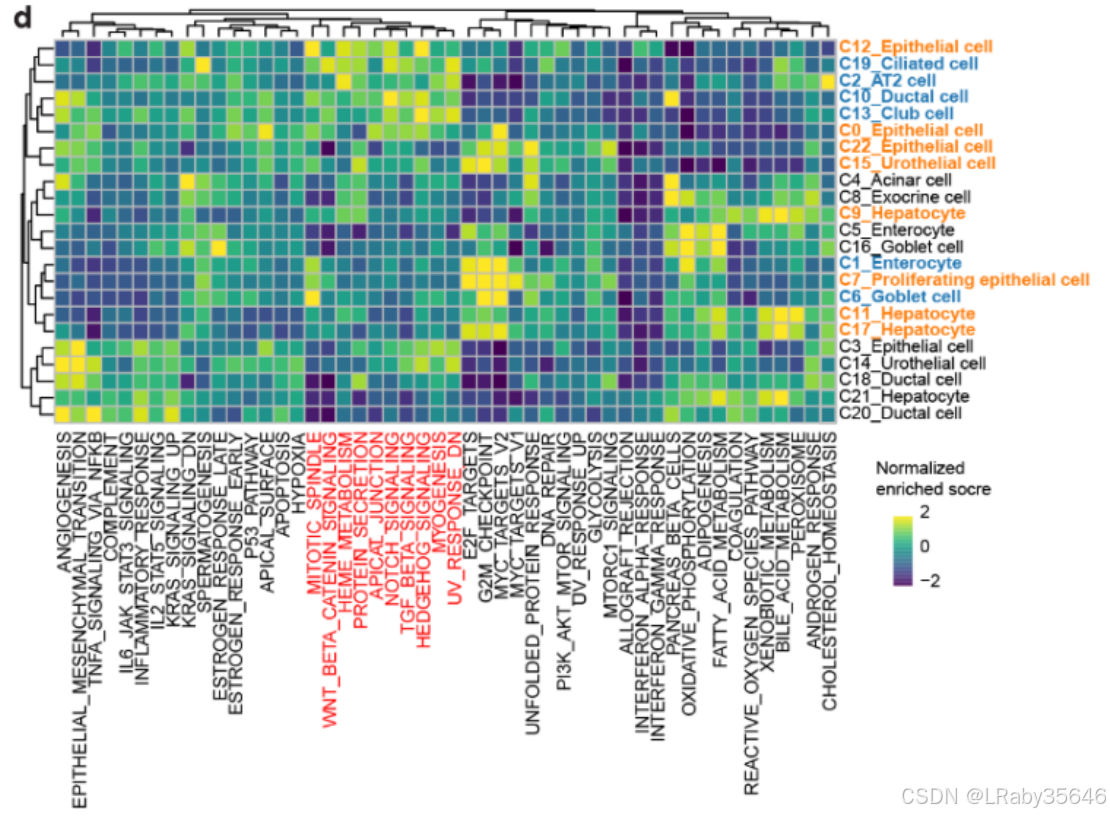
4. 肿瘤发育中的调控模式
对于 肿瘤发展中的调控模式,关注了可能参与调控的 regulon(调控子)(Fig 3d)。
-
恶性集群 和 正常集群 展示了明显不同的调控模式。
-
癌前集群 显示出与 恶性 和 正常 集群的共享调控模式。
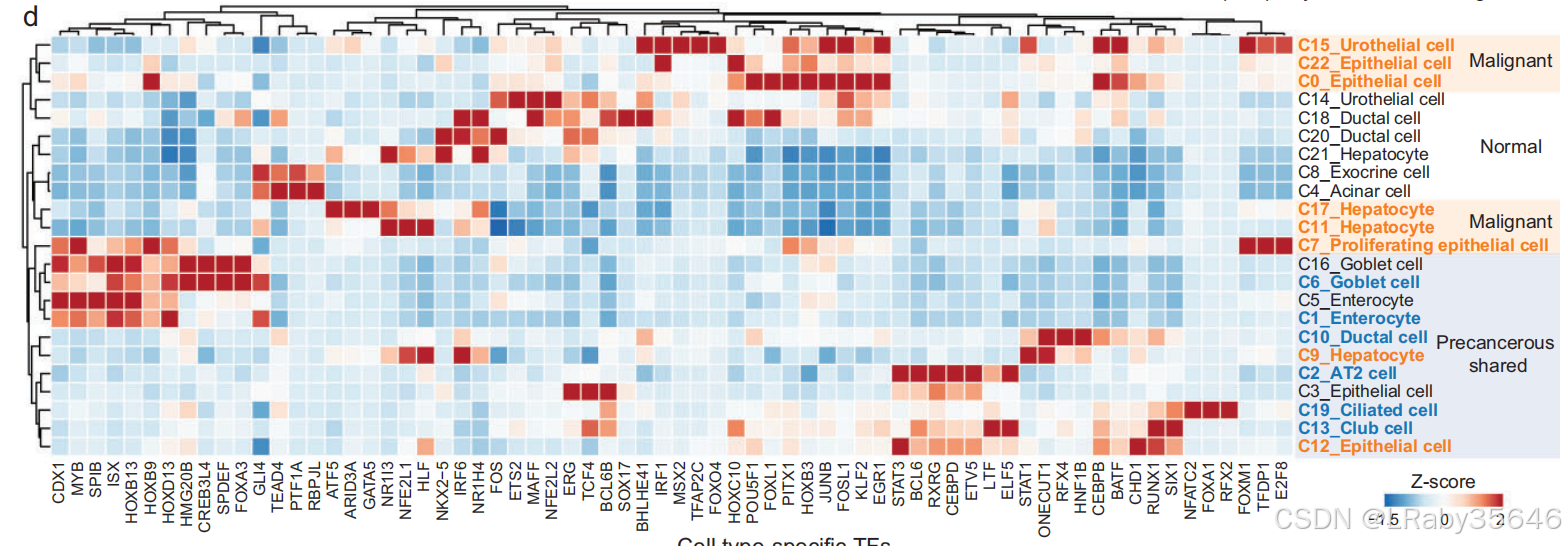
5. 调控因子分析(Fig 3e)
-
恶性集群 富集了与 肿瘤发生和进展 相关的调控因子,例如 EGR1、FOSL、BATF、HOXB3。
-
正常集群 富集了与 细胞发育和分化 相关的调控因子,如 TCF4。
-
癌前集群 显示出与肿瘤进展相关的调控因子,包括:
-
与 肿瘤增殖和细胞周期进程 相关的转录因子(如 STAT1、BCL6、ETV5)。
-
潜在的 肿瘤抑制因子(如 ELF5、LTF、RXRG、CEBPD)。
-
与 上皮-间质转化(EMT) 相关的调控因子。
-
6. 肿瘤转移相关转录因子(Fig 3e)
-
在 肺组织 中收集的细胞(如 C2、C3、C12、C13 和 C19)中,恶性和癌前细胞共享的转录因子包括:
-
LTF、IRX5、SIX1 和 RUNX1,这些因子与肿瘤 侵袭 和 转移 相关。
-
-
潜在的肿瘤抑制因子 ELF5 仅出现在 癌前细胞集群 中。
-
EMT 调控因子 SOX2 和 TCF4 在正常和癌前细胞集群中共享。
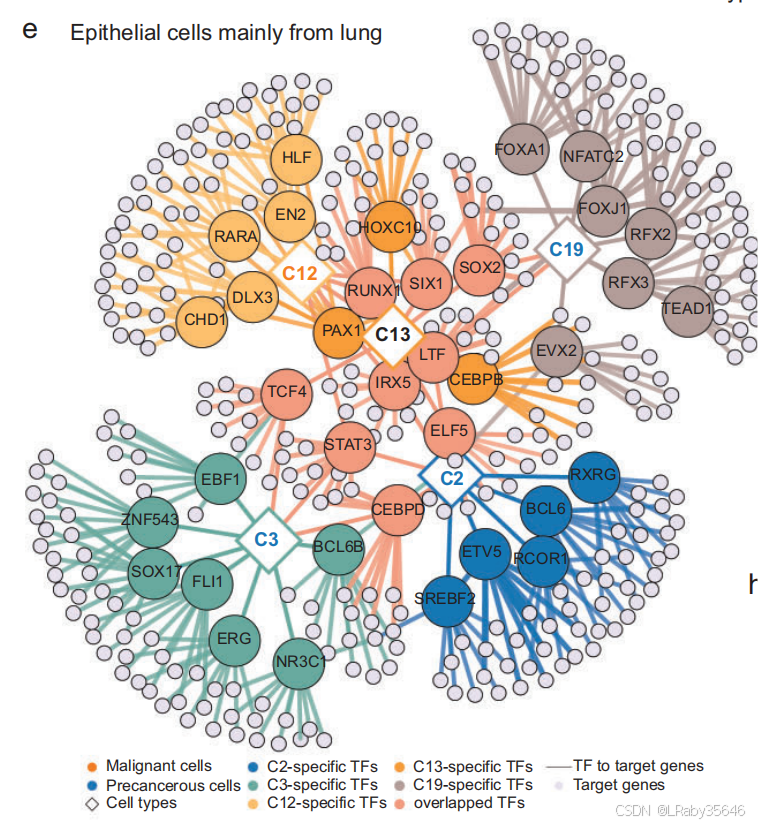
这部分描述了通过 细胞聚类、代谢分析、信号通路分析和调控因子分析,如何揭示 癌前细胞的特征 和 肿瘤发育中的不同调控模式。这些发现强调了 癌前细胞 在肿瘤和正常组织之间的过渡性,突出了 精确识别癌前细胞 的重要性。
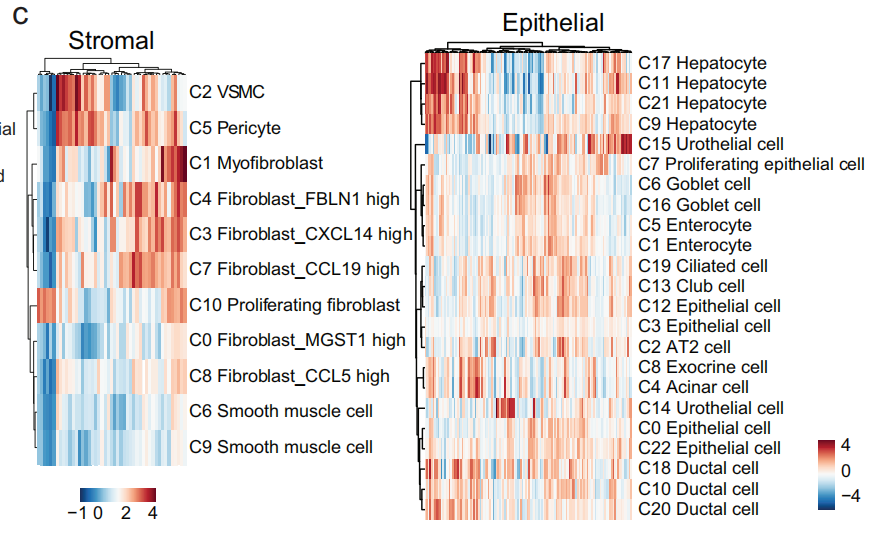
5.3 通过全癌症分析对癌症相关成纤维细胞和内皮细胞的表征
与恶性细胞类似,肿瘤微环境(TME) 的组成异质性是肿瘤的重要组成部分,它与恶性细胞的相互作用显著影响肿瘤的 进展 和 转移
成纤维细胞
1. 细胞间通讯分析
通过 细胞间通讯分析,我们发现 基质细胞、内皮细胞和髓系细胞 在 肿瘤微环境 中的 相互作用最为显著。
-
基质细胞(stromal cells) 与其他组分的 互动最为频繁,无论是肿瘤组织还是邻近组织样本,这种相互作用在 所有组织类型中都显著(所有 p < 0.05)(Fig 2c 和Fig S2d)
2. 基质细胞的表征
-
为了表征 TME 中的 基质细胞,重新聚类了来自每个患者的 所有基质细胞,并将其整合到 全癌症景观 和 HCL 数据中。
-
发现 正常组织 和 肿瘤相关组织 之间的基质细胞存在 明显的分离(Fig 3f 和Fig S7a)
-
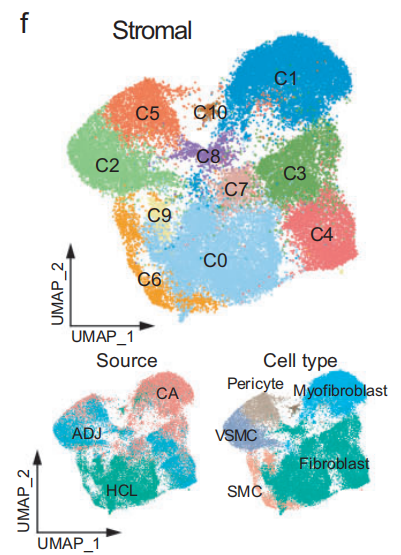
3. 基质细胞的 CNV 分析
-
与 上皮细胞 不同,肿瘤和邻近组织中的 基质细胞 的 CNV 分数 并没有显著差异,但显著高于 HCL 中的正常基质细胞(Fig S7b)。
-
这种 差异不显著 的结果表明,邻近组织中的基质细胞 可能表现出与 肿瘤组织中的基质细胞 相似的特征,这可能与 肿瘤微环境(TME)内的复杂相互作用 有关。
-
4. 基质细胞 在 肿瘤微环境(TME) 中的 聚类特征
35,661 个基质细胞 被划分为 11 个独特的集群(Fig 3f-g,Fig S7c-d)

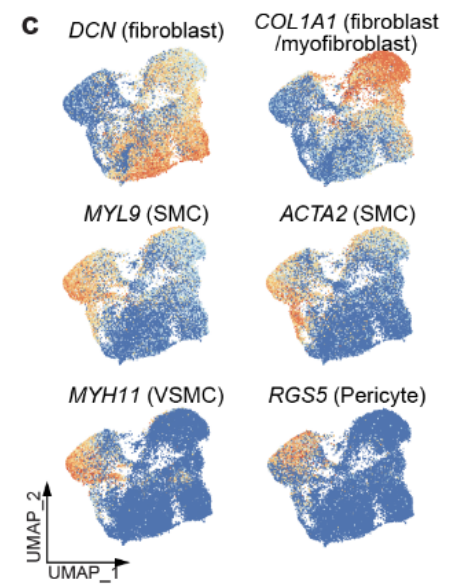
5. Gene Set Enrichment Analysis (GSEA)
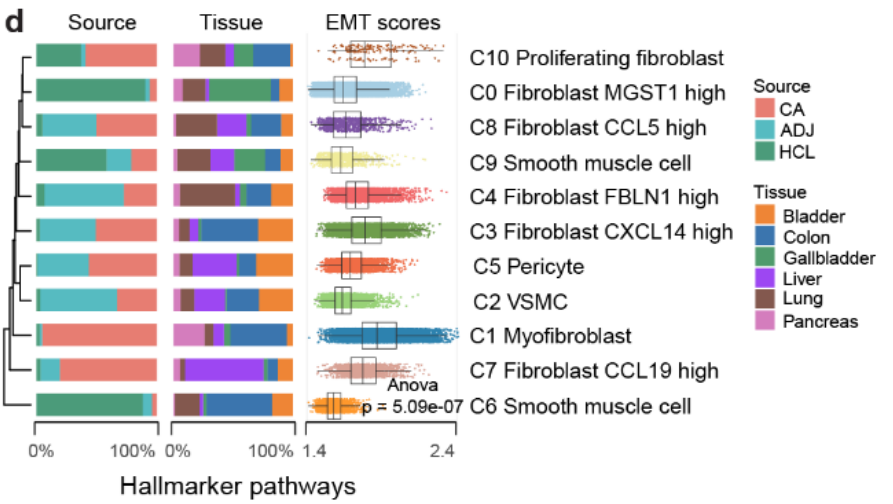
肌成纤维细胞(myofibroblasts)与其他 基质细胞、癌症相关成纤维细胞(CAFs)(如 C3、C4、C7、C8)和 正常成纤维细胞 相比,显示出显著 更高的 EMT 分数(Fig S7d)。
-
这表明 肌成纤维细胞 在 癌症相关成纤维细胞激活过程中 发生了 去分化,并可能是 上皮转分化(epithelial transdifferentiation)过程的主要来源。
-
上皮转分化可能是 肌成纤维细胞 的 主要来源,并为 治疗策略 提供了 新的潜在靶点。
EMT 分数: 是一个用于衡量细胞在 上皮-间质转化(EMT) 过程中的程度的定量指标。EMT 是一种生物学过程,其中上皮细胞失去其上皮特性(如细胞连接和极性),转化为具有间质细胞特征(如迁移性和侵袭性)的细胞。这一过程在 肿瘤发生、转移和组织修复 中起着重要作用。
6. Hallmark 分析结果
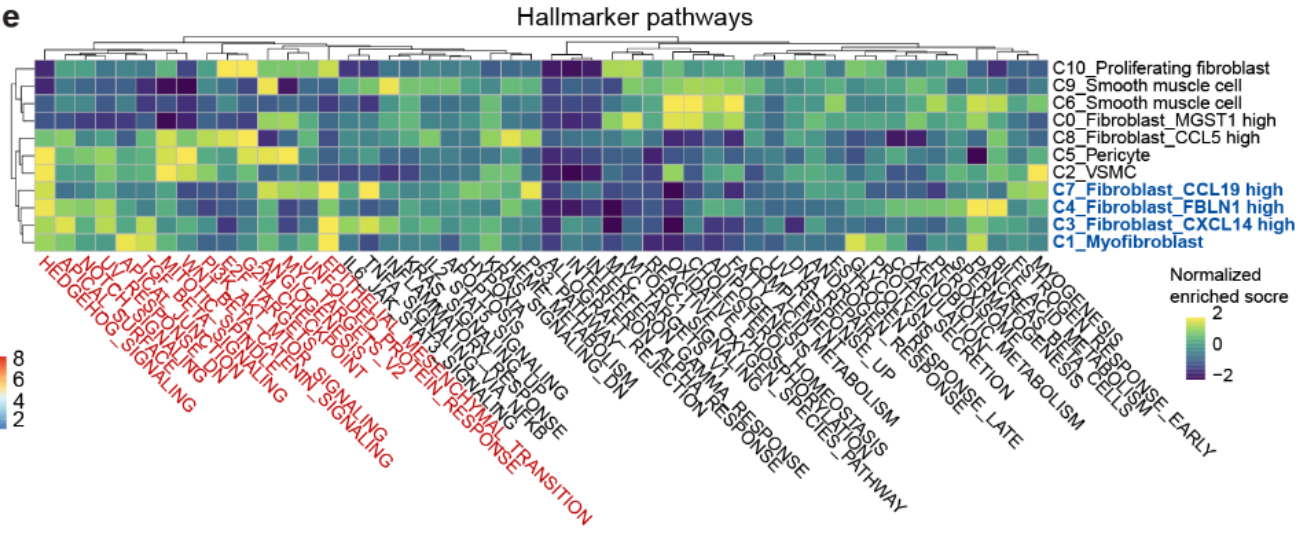
Hallmark 分析 显示,癌症相关成纤维细胞(CAFs)中 氧化磷酸化下调 和 糖酵解信号上调,并富集在与 肿瘤发生和进展 相关的通路中(Fig S7e)。
-
这些结果与 恶性细胞 和 癌前细胞 的分析结果一致(Fig 3b 和Fig S6d)。
Hallmark 分析 是一种基于 Hallmark 基因集 的分析方法,用于识别和理解生物学过程中基因的富集模式。该分析方法通常用于 基因表达数据 中,以确定样本中与已知的关键生物学途径相关的基因的富集情况。
Hallmark 基因集简介
Hallmark 基因集 是 MSigDB中定义的一个标准化基因集,包含了一组代表性强、功能明确的 基因集合,这些基因集合代表了 癌症研究 中常见的 生物学过程、信号通路 或 肿瘤特征。
Hallmark 基因集 中的基因集合包括多种与 癌症发生、发展、转移 和 细胞生物学过程(如 细胞周期、代谢、免疫反应、上皮-间质转化(EMT) 等)相关的通路。
7. CAFs 的调控模式
- 细胞周期 和 细胞增殖相关基因集 在 CAFs(如 C1、C3、C4、C7、C8)、周细胞(C5)和 血管平滑肌细胞(VSMC,C2) 中富集,这些细胞均来自 肿瘤组织和邻近组织。
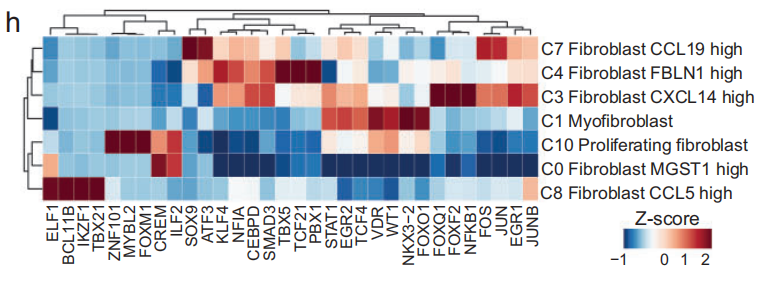
-
在关注 调控模式 时,发现 CAFs 展示了 细胞类型特异性的转录因子(TFs),并且与 广泛参与癌症发生的共享转录因子 一起出现(Fig 3h)。
这些转录因子包括各种 肿瘤促进因子 或 肿瘤抑制因子,如 KLF4、NFIA、TCF21、NFKB1 和 EGR1。 - 这些结果表明,CAFs 可能具有不同的 分化状态,并对 肿瘤微环境(TME) 产生不同的影响,同时分享类似的 调控模式。
内皮细胞
从 全癌症景观 和 HCL 数据 中获得了 17,564 个内皮细胞(Fig S8a–c),并将它们划分为 10 个细胞集群
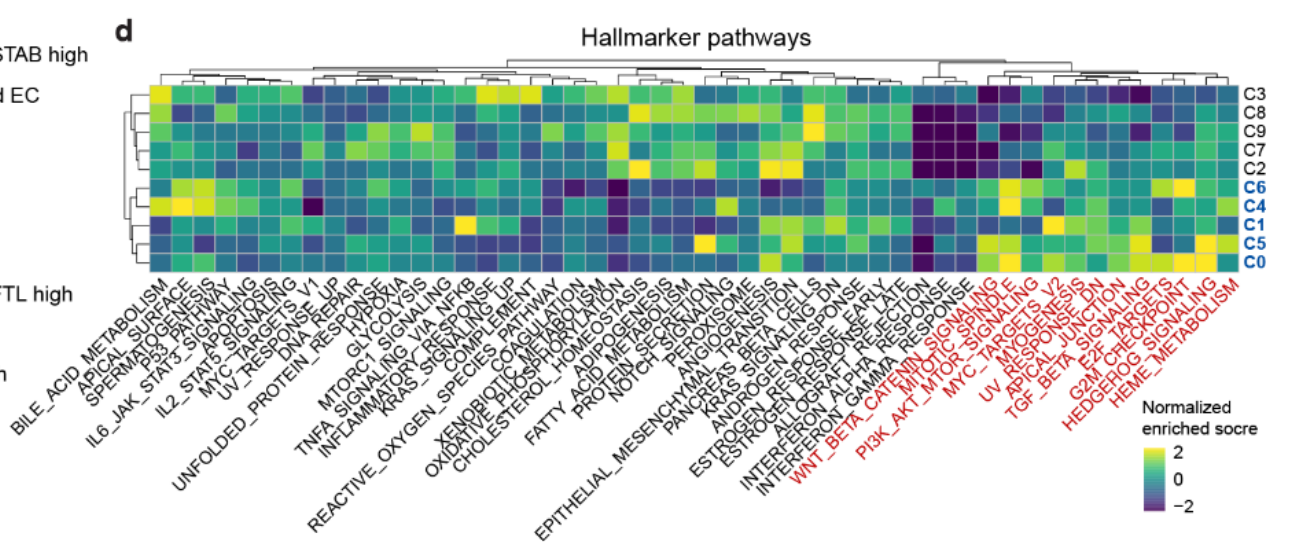
1. 氧化磷酸化下调
揭示了 癌症相关内皮细胞(C0、C1、C4、C5 和 C6)中的 氧化磷酸化下调,这些细胞主要来自 肿瘤组织和邻近组织(Fig S8d)
在这些集群中,与 细胞增殖 和 肿瘤启动 相关的基因集被显著富集,包含以下通路:
-
E2F 目标基因
-
G2M 检查点
-
MYC 目标基因 v2
-
Wnt 信号通路
-
PI3K/AKT/mTOR 信号通路
-
Notch 信号通路
-
TGF-β 信号通路
-
Hedgehog 信号通路
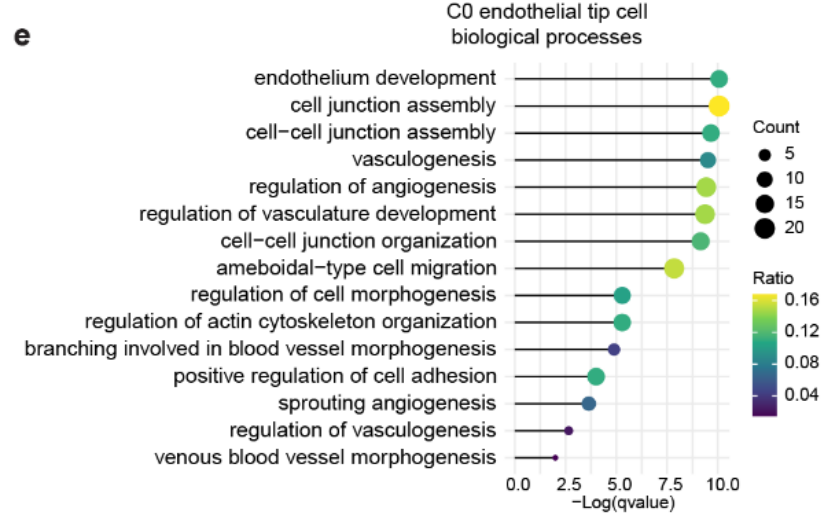
2. C0 集群与血管生成的富集
以 肿瘤组织 为主要来源的 C0(内皮尖端细胞) 集群,在 血管生成调控 相关过程中的 富集程度显著(Fig S8e)。
-
这表明 癌症相关内皮细胞 可能通过 血管生成刺激 参与肿瘤的 重塑和进展。
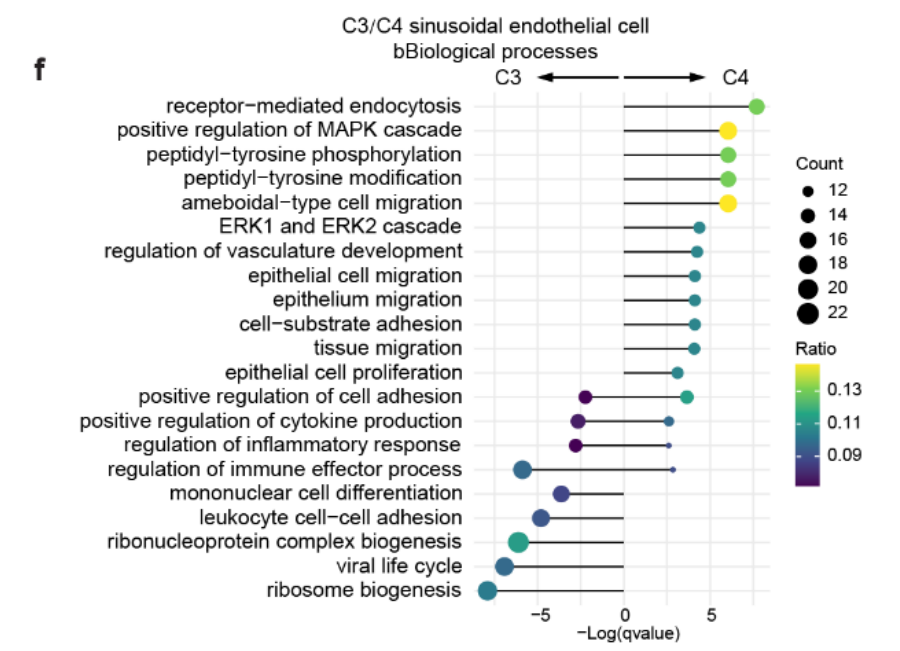
3. 邻近组织中的窦状内皮细胞
来自 邻近组织 的 窦状内皮细胞(C4) 与 正常窦状内皮细胞(C3) 相比,显示出与以下过程相关的显著富集:
-
ERK1 和 ERK2 信号级联
-
上皮细胞迁移和增殖
-
细胞-基质粘附(Fig S8f)
5.4 使用深度学习框架进行可解释的单细胞水平药物扰动预测
深度学习框架‘神农“”’能够描述 个体癌细胞 对药理性扰动的响应,筛选潜在的抗癌药物靶点,并评估可能的组织损伤效应,通过机器学习方法进行 单细胞层面的预测 可以加速 药物发现过程,并提高药物筛选的 准确性 和 效率。
1. Shennong框架的组成
在上面方法部分
2. Shennong框架应用于药物候选筛选
为了发现药物候选物及其潜在靶点,将 Shennong 框架应用于 全癌症景观,探索肿瘤细胞对药理化合物的反应。
-
通过 随机选择约90%的细胞(346,129个细胞)构建了 训练集,其余 42,517个细胞 作为 预测集,用来评估 Shennong 是否能正确研究单细胞扰动的效果。
-
训练集和预测集都包含来自 肿瘤、邻近 和 健康正常组织 的细胞,涵盖 上皮、内皮、淋巴、髓系 和 基质 细胞谱系。
-
成功的学习应当能够提取来自不同组织来源的细胞的 独特 和 共同特征,并将这些细胞映射到不同的细胞类型,同时计算术语的强效应。
-
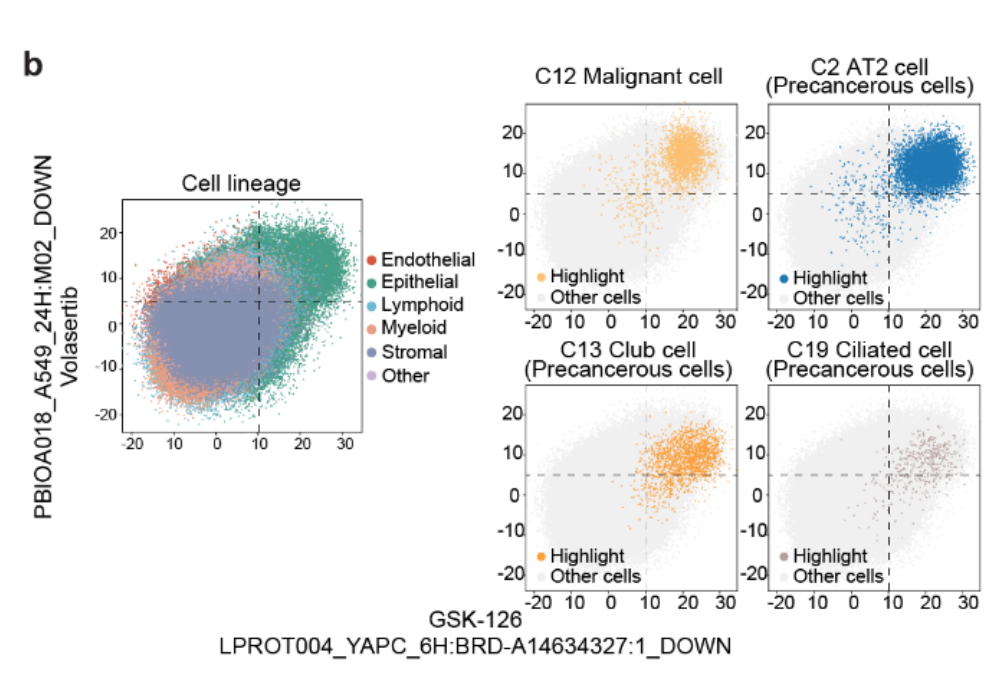
经过训练和预测后,成功地从训练集中提取特征,并将这些特征映射到预测集中(Fig 4b 和Fig S9b–d)
-
预测集中的细胞被分成不同的簇,且相同谱系的簇倾向于聚集在一起(Fig 4b)
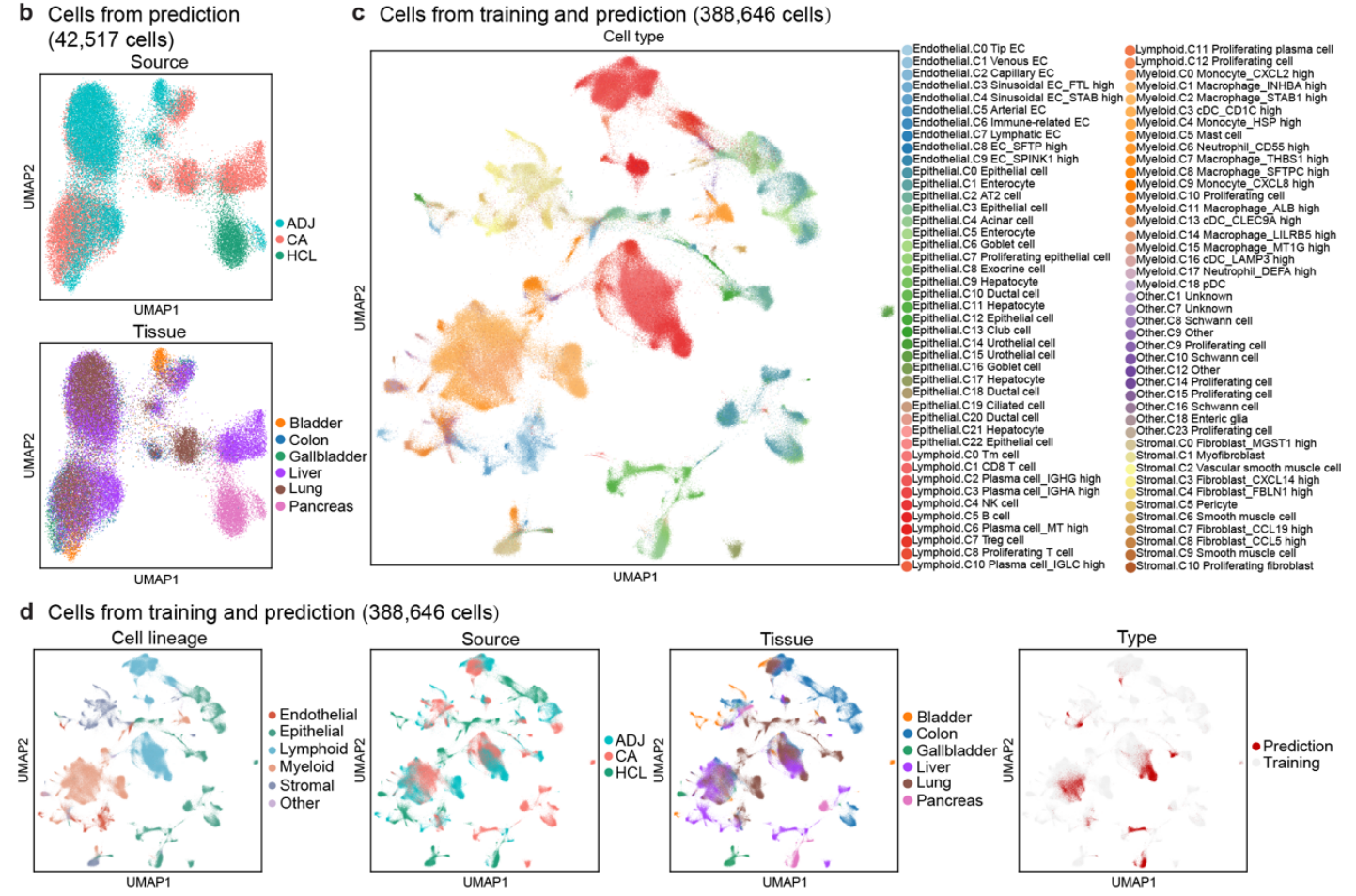
-
预测细胞的聚类和分布与 单谱系分析 中的 细胞类型注释 一致(Fig 3a, f 和Fig S8a)。
-
这些簇主要来自 单一组织来源 或 组织类型(例如上皮细胞),而其他谱系的簇则跨越不同的组织来源和组织类型(Fig S9b)
-
进行了 10折交叉验证分析,结果表明特征提取和细胞聚类的结果高度可重复,证明了 Shennong框架 的稳健性(Fig S10)
- 基于每个细胞的 影响术语分数,识别了每种细胞类型中显著差异的术语,并使用 贝叶斯因子 进行统计(表14)
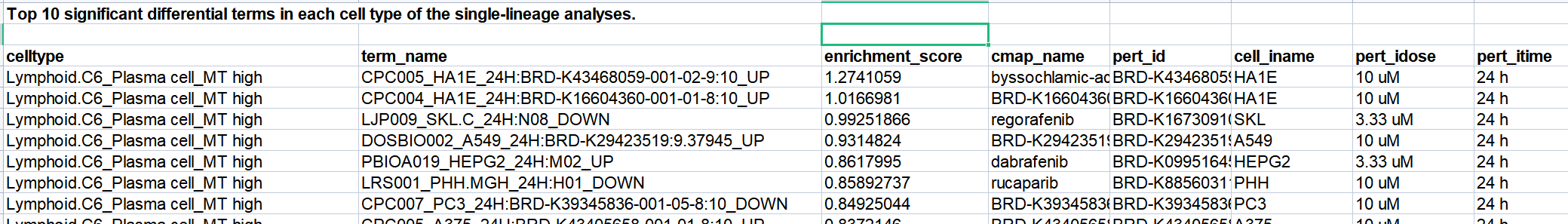
- 使用每种细胞类型中的 前10个显著差异术语,能够在 单谱系分析 中区分不同的细胞类型(Fig 4c 和Fig S11a, b)。
- 通过计算这些术语,发现一些术语在多个细胞类型中出现(Fig 4d,标记了对应的化合物),这些化合物已在许多癌症类型中进行了大量实验或临床试验。
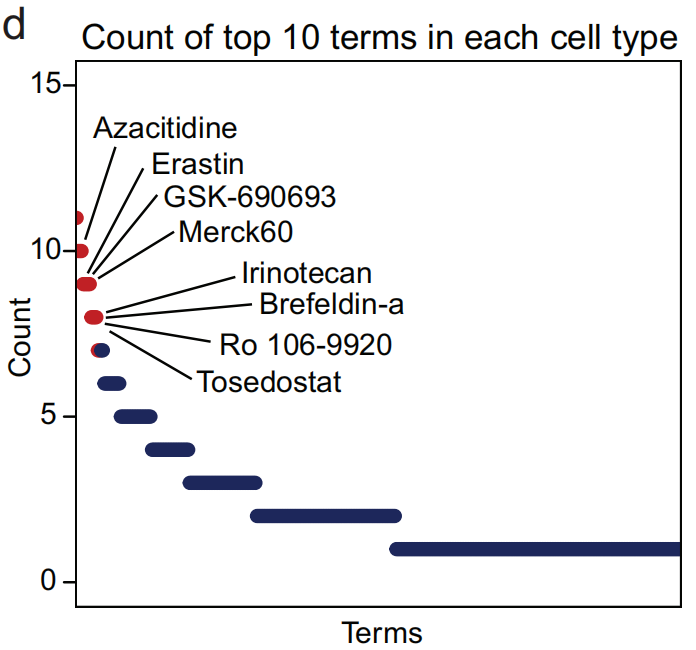
- 已批准的药物示例 (阿扎胞苷(Azacitidine),伊立替康(Irinotecan),托塞多STAT(Tosedostat)
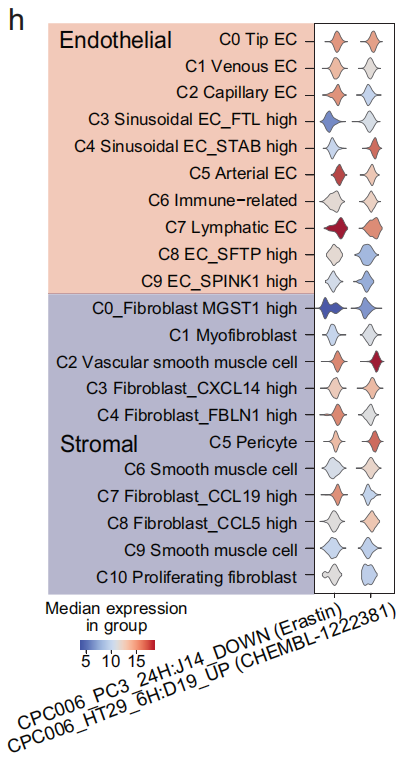
- CAF(癌症相关成纤维细胞)(C1、C3、C4 和 C7)在 扰动影响 上表现出与 正常成纤维细胞 和 平滑肌细胞 不同的特征(Fig 4c),类似的结果也出现在 内皮细胞 中(Fig S11b)
-
通过计算每个术语的 富集分数,发现 LJP008_SKL_24H: G19_DOWN 和 MOA001_U2OS_24H: P04_DOWN 是最常见的显著差异术语(Fig 4e),对应于从 阿扎胞苷 和 帕尔班达唑(Palbendazole) 提取的显著特征。
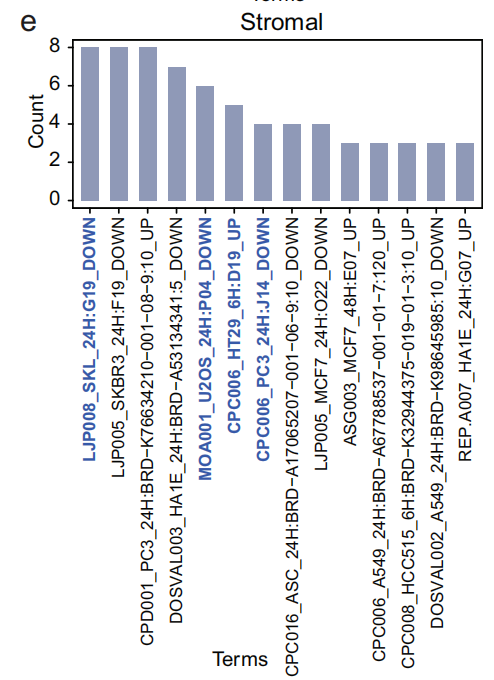
- 这些术语在 CAF 中富集,而不是在其他基质细胞或其他谱系的细胞中(Fig 4f),并能将 CAF 从其他细胞中分离开来(Fig 4g)。
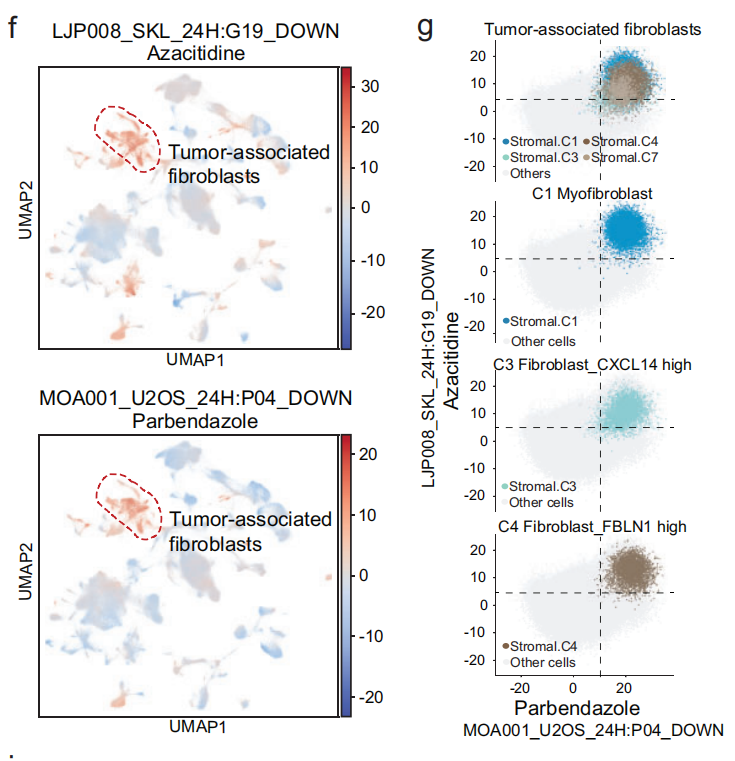
- 这表明 阿扎胞苷 和 帕尔班达唑 对 CAF(由来自多个组织的细胞组成)非常敏感,并可能具有 泛癌症治疗 的潜力。
-
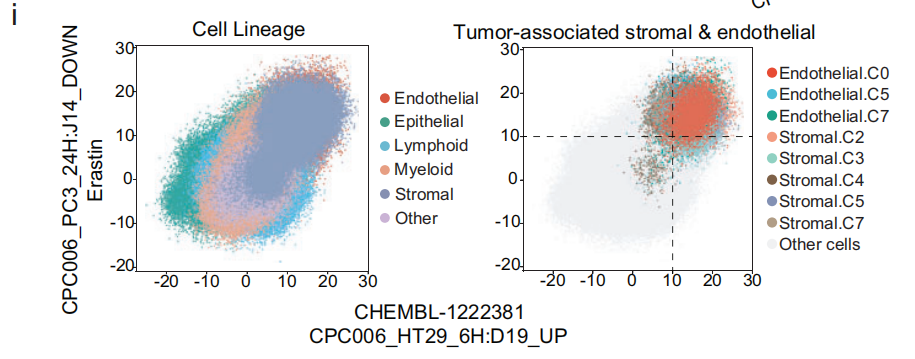
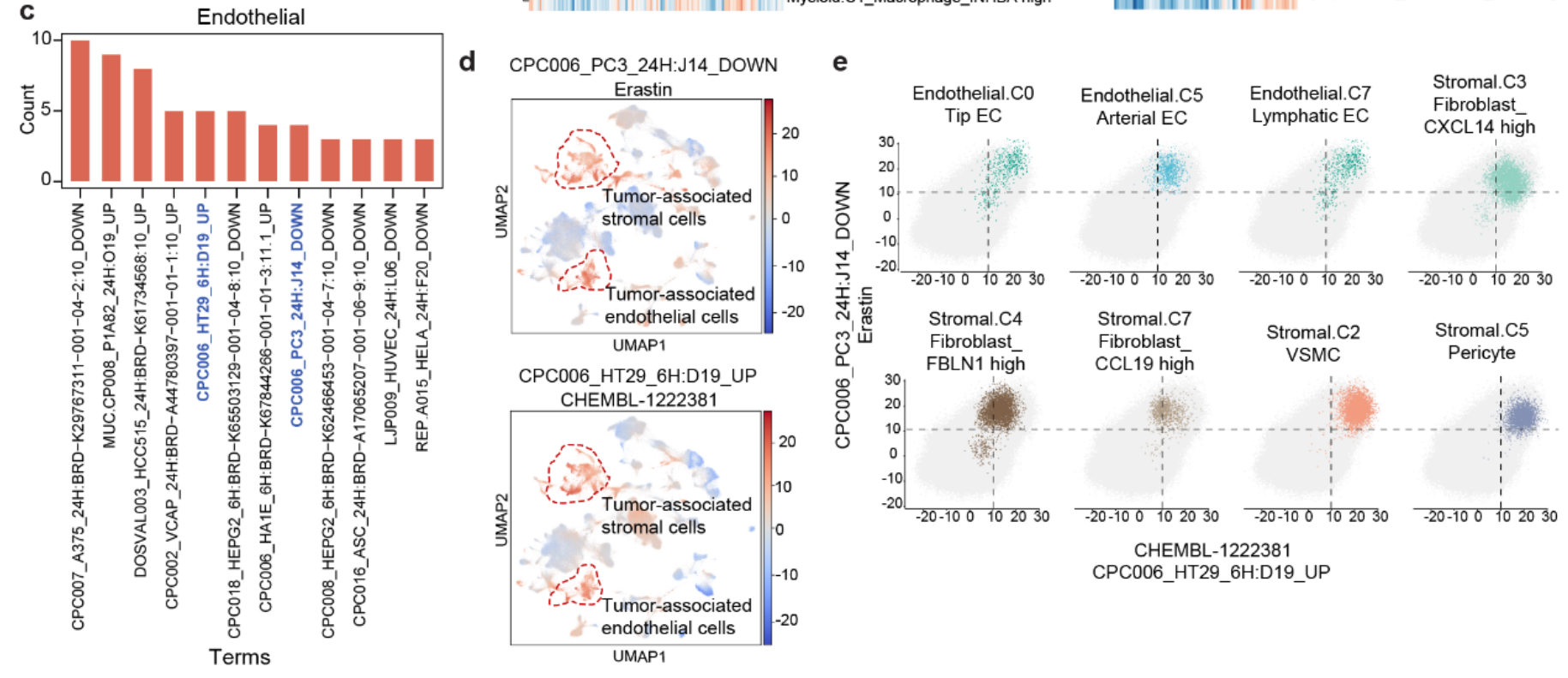
还发现 CPC006_HT29_6H: D19_UP 和 CPC006_PC3_24H: J14_DOWN 在 基质细胞 和 内皮细胞 中出现得较为频繁(Fig 4e 和 S11c),并且在 癌症相关基质细胞 和 内皮细胞 中尤其富集(Fig4h 和 S11d)。这些两种术语能够通过富集 癌症相关基质细胞 和 内皮细胞,将其从 正常基质细胞 和 内皮细胞 或其他谱系的细胞中分离出来(Fig 4i 和 S11e)。
5.5 使用 Shennong 识别抗癌药物及其潜在靶点
1. CAFs与肿瘤细胞的组织特异性
-
与来自不同组织的 CAF 不同,大多数 恶性细胞 和 癌前细胞 是组织特异性的。
-
每种上皮细胞类型中的 显著差异术语 是不同的,但一些术语在主要来自相同组织的细胞类型中会重复出现(Fig S12a)
2. 肺部恶性细胞和癌前细胞中的富集术语
-
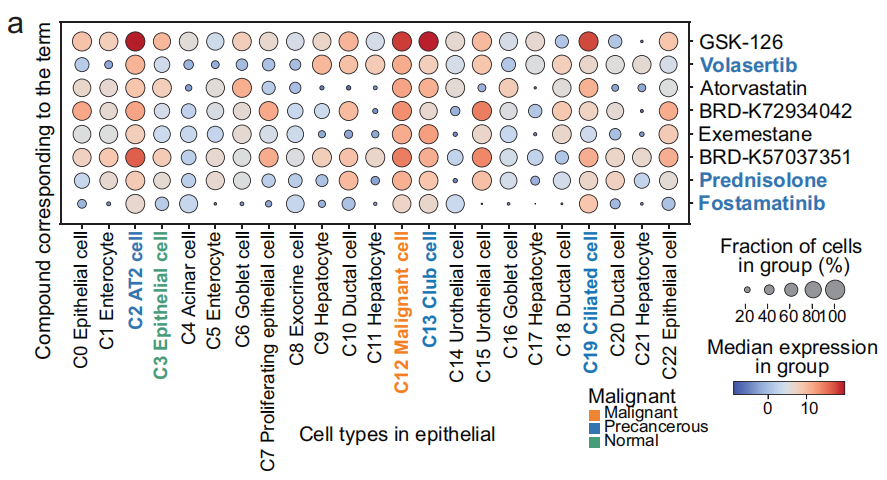
一系列术语在 肺部恶性细胞 和 癌前细胞 中显著富集,但在 正常肺上皮细胞 中没有显著富集(Fig 5a)
3. 相关化合物
-
这些术语对应的化合物包括 FDA 批准药物、实验药物 和 实验化合物,其中大多数已被证明能够抑制 肿瘤生长 和 肿瘤扩展,特别是在 肺癌 中。
-
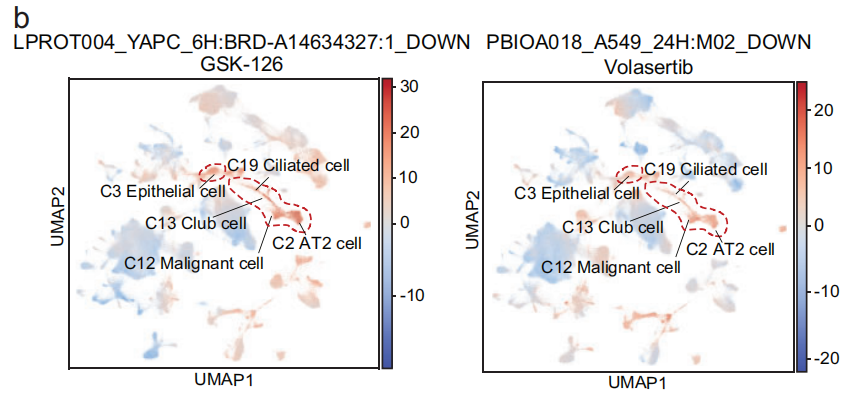
例如,术语 LPROT004_YAPC_6H: BRD-A14634327:1_DOWN 和 PBIOA018_A549_24H: M02_DOWN 在 恶性细胞 和 癌前细胞 中富集,包括 AT2 细胞(C2)、俱乐部细胞(C13)和 纤毛细胞(C19),对应的化合物分别是 GSK-126 和 volasertib(Fig 5b)。使用这两个术语,可以将来自 肺部的恶性细胞 和 癌前细胞 区分(Fig 5c 和 S12b),相比之下,这些术语在 正常肺上皮细胞 和其他组织的 上皮细胞 中的影响分数较低,表明相应的化合物对这些细胞不敏感。
-
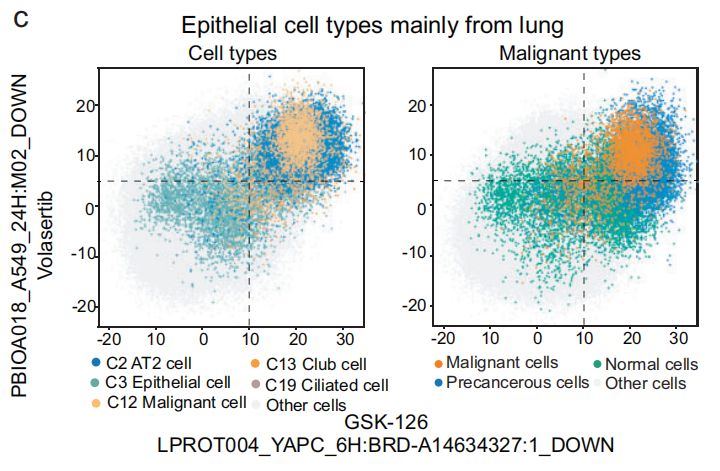
- 下面介绍其他药物的应用(Fig 5d)

4. 分析术语中基因的贡献
- 为了研究药物的作用机制,分析了每个术语中的 个别基因贡献,以阐明相应化合物的潜在靶点和相关信号通路。
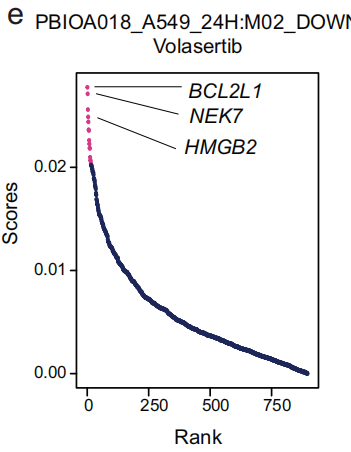
- 以Volasertib为例:Volasertib 可以导致 BCL2L1 表达下调,破坏 PLK1 和 NEK7 之间的相互作用,或将 HMGB2 从有丝分裂染色体中置换,从而影响肿瘤细胞的有丝分裂停滞和凋亡。这些基因是 术语 PBIOA018_A549_24H: M02_DOWN 的重要贡献者(Fig 5e),该术语对应于 小分子抑制剂药物 Volasertib(对 Polo-like 激酶1,PLK1)。
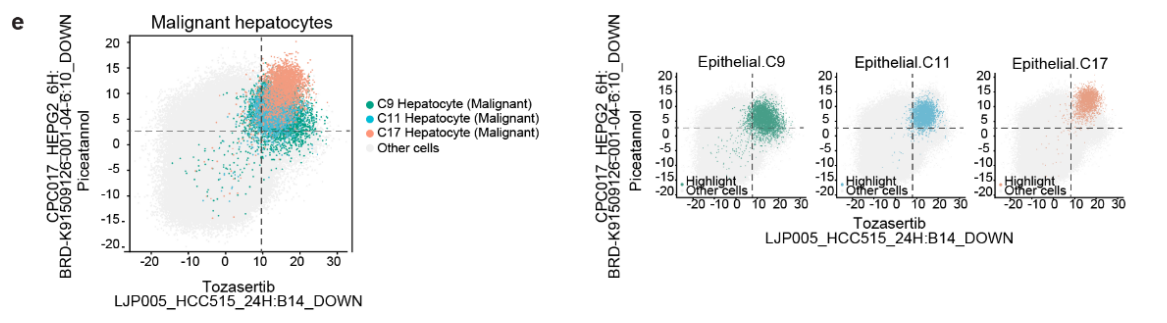
5. 关注肝脏恶性细胞的术语
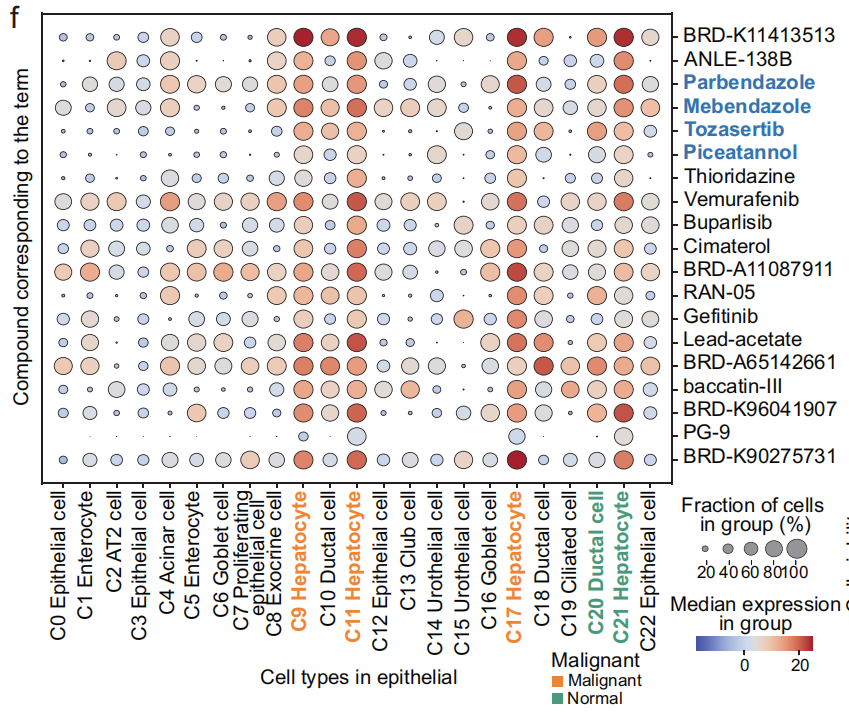
- 收集了 肝脏恶性肝细胞 中 前10个显著差异术语(Fig 5f)。
-
大多数术语在 恶性肝细胞 中显著富集,但在 正常肝细胞 中没有显著富集,术语包括:
-
PBIOA019_HEPG2_24H: M19_DOWN
-
CPC002_HCC515_24H: BRD-K77987382-001-08–2:10_DOWN
-
LJP005_HCC515_24H: B14_DOWN
-
CPC017_HEPG2_6H: BRD-K91509126-001-04–6:10_DOWN(Fig 5g 和 S12c)
-
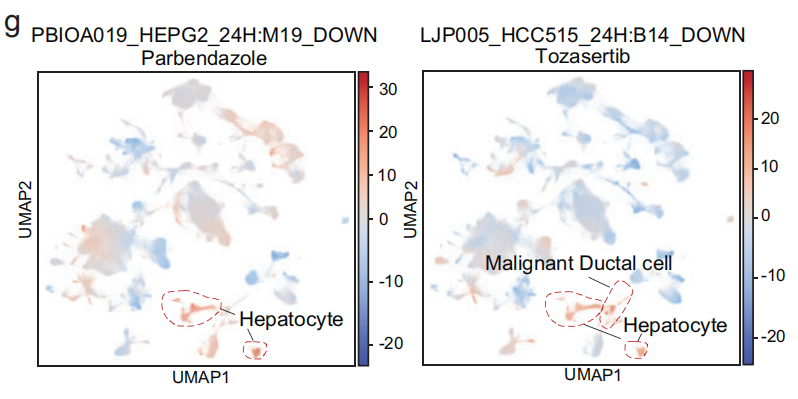
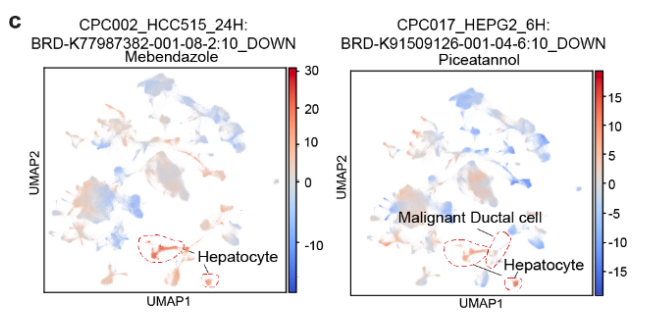
- 这些术语对应的化合物包括 帕尔班达唑、美班达唑、托萨塞替布 和 皮克塔诺洛。
- 使用这四个术语,肝脏恶性肝细胞 可以与其他细胞区分开来(Fig S12d, e),这暗示了这些化合物在 肝脏肿瘤 中的 抗癌活性。
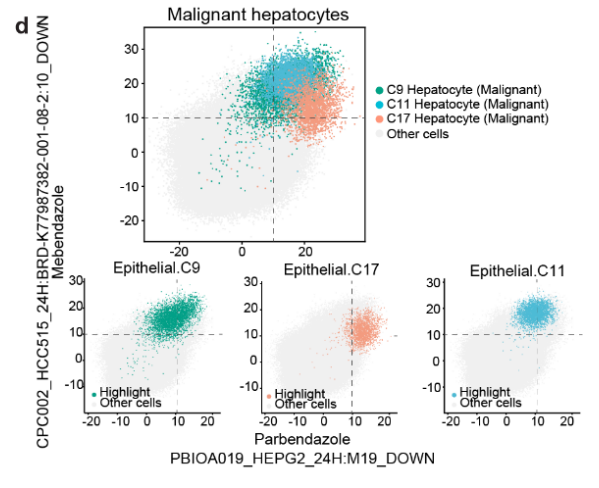
- 讨论4种药的抗癌活性
在HepG2细胞系中的筛选
-
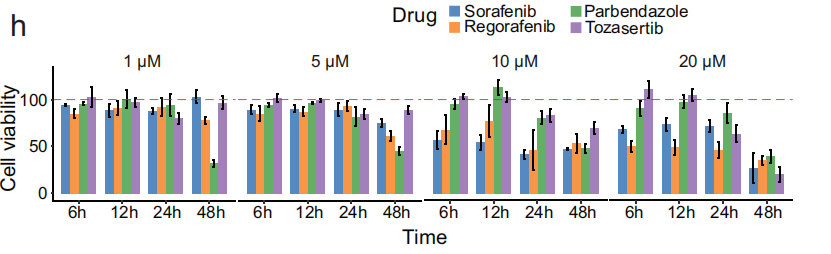
对 HepG2 细胞系 进行的 细胞筛选 确认了 帕尔班达唑 对 肝癌 的 抗癌活性(Fig 5h),其效果可与治疗 晚期肝细胞癌(HCC) 的标准疗法(如 索拉非尼 和 瑞戈非尼)相媲美。
6. Shennong框架 在不同数据集中的 预测结果比较
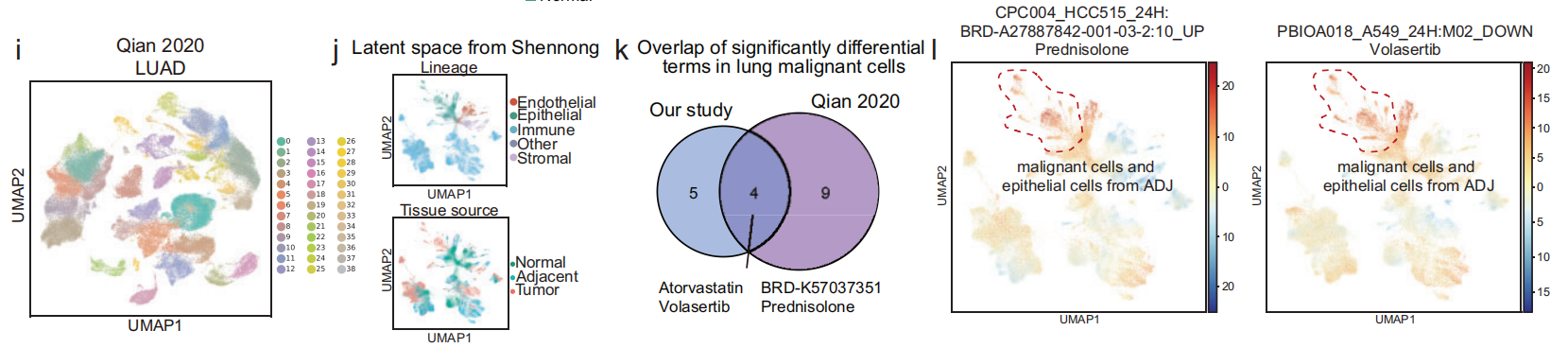
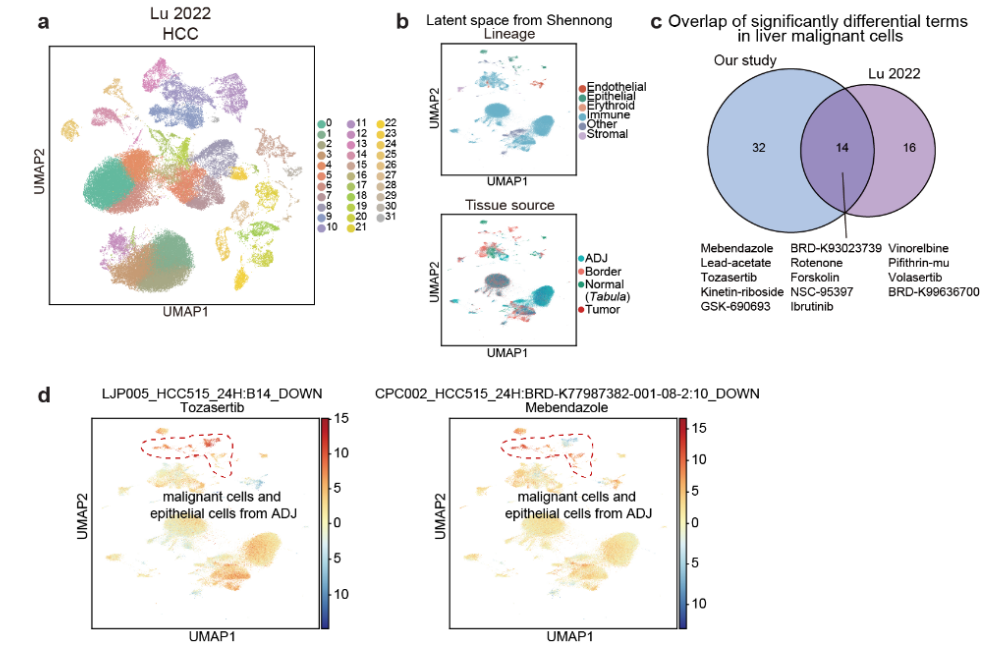
- 为了增强框架的 鲁棒性,将泛癌症景观(即 恶性细胞的显著术语)的预测结果与 第三方数据集 进行了比较(Fig 5 i–l 和 S13a–d)。
模型的特征提取
-
训练后的模型成功提取了这些数据集中来自不同组织来源的细胞的 独特和共同特征,并且潜在空间显示了来自相同谱系的簇在 Shennong框架的预测后 倾向于聚集在一起(Fig 5j 和 S13b)。
重叠的显著差异术语
-
30%∼45% 来自泛癌症景观的 恶性细胞 中的显著差异术语与对应的 第三方数据集 中的术语重叠(Fig 5k 和 S13c)。
-
只有在至少 两个簇 中观察到的术语才被计算在内。
-
重叠的化合物
-
这些重叠的术语包括 实验药物 volasertib 和 tozasertib(均在临床试验中),以及新的候选药物 泼尼松龙 和 美班达唑(Fig 5k 和 S13c)
5.6 使用 Shennong 评估抗癌药物的组织损伤效应
1. 预测抗癌药物的副作用
- 当关注在 肿瘤相关基质细胞 和 内皮细胞 中富集的术语时,发现 CPC006_PC3_24H: J14_DOWN 也在 正常内皮细胞(C2 和 C7)中富集(Fig 4h)。这表明与该术语相关的小分子化合物可能具有 组织损伤效应。Erastin,对应 CPC006_PC3_24H: J14_DOWN,在 前临床研究 中已显示对健康组织具有毒性,特别是在 铁死亡 诱导方面。
2. Shennong框架 预测药物对 正常细胞的潜在损伤效应
- 一些术语不仅在 恶性肝细胞 中富集,而且在主要来源于 HCL 的细胞类型中也富集。
- 这表明与这些术语相关的小分子化合物也可能对 正常细胞 敏感。
-
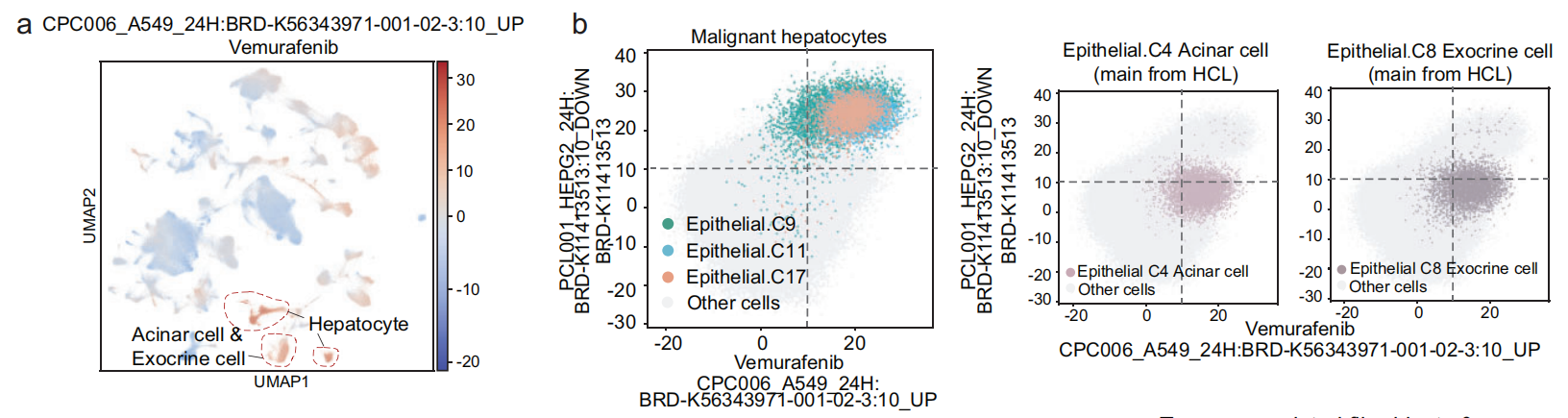
以CPC006_A549_24H为例
-
例如,术语 CPC006_A549_24H: BRD-K56343971-001-02–3:10_UP 在 肝细胞、腺泡细胞(C4)和 外分泌细胞(C8)中有显著的影响力术语得分(Fig 6a, b)。
-
与 恶性肝细胞 不同,腺泡细胞 和 外分泌细胞 来源于 正常胰腺组织,这表明相关化合物可能对正常的胰腺上皮细胞具有 组织损伤效应。该术语对应的化合物 Vemurafenib 是一种 BRAF抑制剂,用于治疗 Erdheim-Chester病 和 黑色素瘤 的靶向治疗。有报道显示,使用 Vemurafenib(Zelboraf)与 胰腺炎 相关。
-
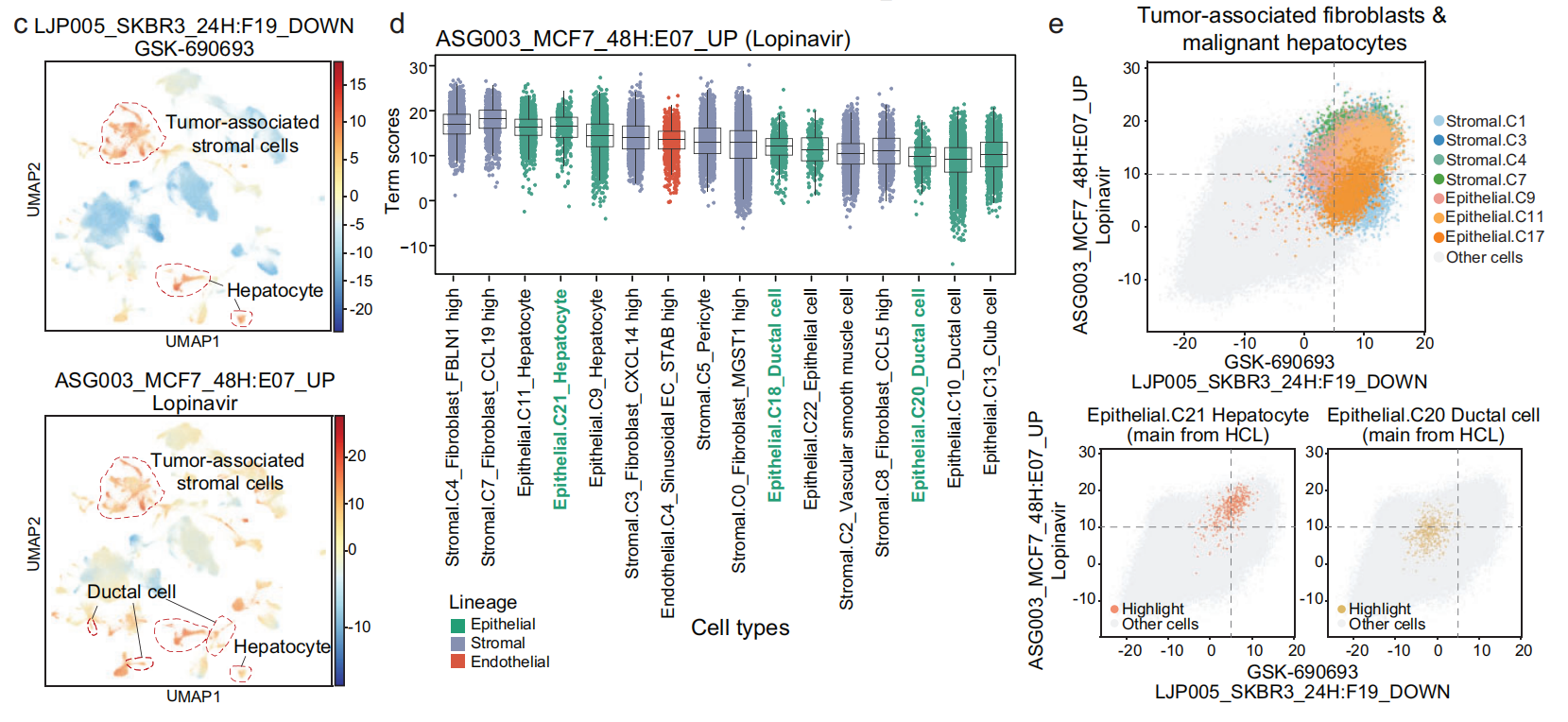
- 在 基质细胞 和 内皮细胞 中,LJP005_SKBR3_24H: F19_DOWN 和 ASG003_MCF7_48H: E07_UP 是 显著差异术语,并且这两个术语在不同细胞类型中 反复出现(Fig 4e 和 S11c)。
除了 肿瘤相关基质细胞 和 内皮细胞 外,这两个术语在 恶性细胞 和 癌前细胞 中也显示出显著的影响力术语得分,包括:
-
正常成纤维细胞(如 纤维母细胞_MGST1高,基质,C0)
-
肝细胞(上皮,C21)
-
导管细胞(上皮,C18 和 C20)(Fig 6c–e 和 S14a, b)。
这表明与这两个术语相关的化合物对 正常肝上皮细胞 和 基质细胞 具有 显著影响。

6. 局限性
-
扰动数据的依赖性 是本研究的一个关键问题,CMap数据主要提供了具有实验评估转录组的分子。
-
不同数据收集平台可能引入批次效应,且 扰动数据 来自 整体RNA测序,无法捕捉对特定细胞类型的影响,这可能影响结果的 广泛适用性。
-
框架专注于 显著特征,如果扰动数据集不够全面,可能会限制其应用。
-
未来框架的改进可以通过 更大、更丰富的扰动数据集,包括 配体 和 siRNA 来提升预测精度。
-
许多常见副作用表现为全身反应,如 疲劳、脱发、过敏反应、恶心 和 贫血。
-
在分析中,组织损伤效应 主要聚焦于特定细胞类型的 细胞毒性效应,因此框架对于 副作用的预测 可能只部分有效。
-
虽然结果得到了 细胞实验、第三方数据集对比 和 近期临床数据 的支持,但仍然需要 实验验证。
-
后续研究需要 体外 或 动物模型实验 来确认预测的生物学效应、安全性和有效性。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









