






很久之后,关于featurecounts得出的基因长度问题:
python 学习之 featureCounts 软件的基因长度是怎么算的?
但算fkpm、tpm就是用的外显子长度。
-
T2T基因组的RNA-seq数据
https://www.ncbi.nlm.nih.gov/sra/SRX11364663[accn]
https://www.ncbi.nlm.nih.gov/sra/SRX11364664[accn]
一个样本,两个技术重复
另一个来源:
https://github.com/marbl/CHM13/blob/master/Sequencing_data.md
下载fastq.gz文件
https://www.ebi.ac.uk/ena/browser/text-search -
处理SRA数据
ploy(A) plus RNA-seq: 只关心编码区,对于mRNA进行测序
步骤参考:
RNA-Seq:从fastq到表达矩阵
得到表达矩阵的流程
#转换成fastq文件
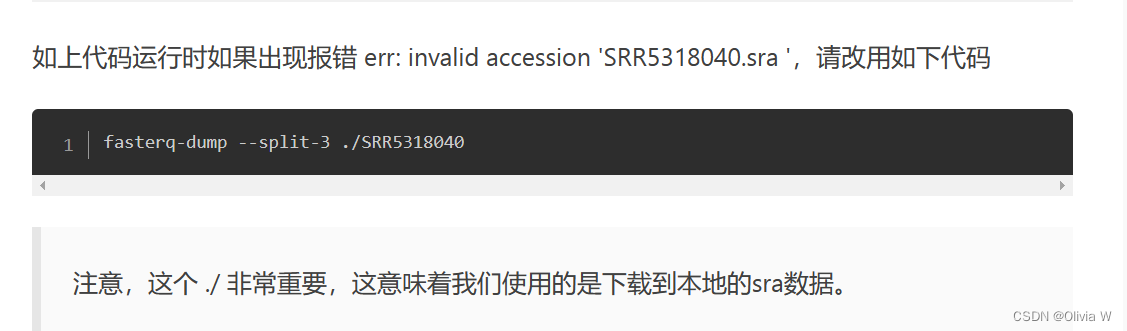
fasterq-dump --split-3 ./SRR15054301
#压缩fastq文件
gzip SRR15054302_1.fastq
Linux运行shell脚本提示No such file or directory错误的解决办法
linux conda install出现condahttperror: http 403 forbidden for url。。。
#fastq文件质控
for file in `ls *_1.fastq.gz | perl -lpe 's/_1.fastq.gz//'`
do
fastp -i ${file}_1.fastq.gz -I ${file}_2.fastq.gz -o ${file}_1.clean.fastq.gz -O ${file}_2.clean.fastq.gz
done
nohup bash qc.sh &
#RNA-seq比对
#1 建立索引
vim hisat2-build.sh
genome=./T2T.fasta
genome_prefix=./T2T
hisat2-build -p 4 $genome $genome_prefix 1>hisat2-build.log 2>hisat2-build.err
#运行hisat2-build.sh
nohup bash hisat2-build.sh &
建立基因组索引只需3、4个线程
map需要32个线程(服务器一个节点是32核)
#比对结果压缩、排序及构建索引
多线程
for file in *.sam
do
samtools view -b $file -@ 8 > /share/home/weirui/SRA_data/sorted/${file%.sam}.bam
samtools sort /share/home/weirui/SRA_data/sorted${file%.sam}.bam -@ 16 > /share/home/weirui/SRA_data/sorted/${file%.sam}.sorted.bam
samtools index /share/home/weirui/SRA_data/sorted/${file%.sam}.sorted.bam
done
#featureCounts定量
线程不要设置太多,比如32,因为有merge步骤合并文件,cpu占用率很低。当我们想对meta-feature水平进行统计的时候,不能设置-f参数
两个技术重复,一个8小时,一个11小时。
featureCounts -p -T 16 -t exon -g gene_id -a /share/home/weirui/SRA_data/references/T2T.gtf -o ./counts.txt ./*.sorted.bam
插个楼:想要gff换gtf gffread不好使会遗漏信息,但gene数量好似没变;agat,试试,一直安装不了
学院的服务器conda按学院的服务器手册网址换源,终于可以了
运行之后其实还是有信息丢失,但是应该无论gtf还是gff关于gene和exon的信息应该一样吧,反正我暂时放弃这步,后续出问题再改正。
最后,代码情况:

main.sh
#cd sra-data
#bash sra_fastq.sh
#cd ..
cd data
bash qc.sh
cd ..
cd hisat2
bash hisat2-run.sh
bash sorted.sh
cd ..
cd sorted
bash featureCount.sh
hisat2-build.sh
genome=./T2T.fasta
genome_prefix=./T2T
hisat2-build -p 3 $genome $genome_prefix 1>hisat2-build.log 2>hisat2-build.err
sra_fastq.sh
for file in SRR*
do
fasterq-dump --split-3 ./$file --outdir /share/home/weirui/rna-seq/data -e 10
done
qc.sh
for file in *.fastq
do
pigz -p 16 $file
done
for file in `ls *_1.fastq.gz | perl -lpe 's/_1.fastq.gz//'`
do
fastp -w 10 -i ${file}_1.fastq.gz -I ${file}_2.fastq.gz -o ${file}_1.clean.fastq.gz -O ${file}_2.clean.fastq.gz -h ${file}.html -j ${file}.json
done
#局限:双端
hisat2-run.sh
genome_prefix=/share/home/weirui/rna-seq/references/T2T
#局限:for paired -end, PE
for sample in `ls /share/home/weirui/rna-seq/data/*_1.clean.fastq.gz | perl -lpe 's/_1.clean.fastq.gz//'`
do
hisat2 --new-summary -p 32 -x $genome_prefix -1 ${sample}_1.clean.fastq.gz -2 ${sample}_2.clean.fastq.gz -S /share/home/weirui/rna-seq/hisat2/${sample:32}.sam 2> /share/home/weirui/rna-seq/hisat2/${sample:32}.err
done
sorted.sh
for file in *.sam
do
samtools view -b $file -@ 8 > /share/home/weirui/rna-seq/sorted/${file%.sam}.bam
samtools sort /share/home/weirui/rna-seq/sorted/${file%.sam}.bam -@ 16 > /share/home/weirui/rna-seq/sorted/${file%.sam}.sorted.bam
samtools index /share/home/weirui/rna-seq/sorted/${file%.sam}.sorted.bam
done
featureCount.sh
featureCounts -p -T 16 -t exon -g gene_id -a /share/home/weirui/rna-seq/references/T2T.gtf -o /share/home/weirui/rna-seq/featurecounts/counts.txt ./*.sorted.bam
count2tpm.ipynb
import numpy as np
np.set_printoptions(suppress=True)
usecols=np.arange(5,43,1)
counts = np.loadtxt('./counts.txt',dtype=float,delimiter='\t',usecols=usecols,skiprows=2)
counts
#%%
for i in range(len(counts)):
for j in range(1,len(counts[0]),1):
counts[i][j]=counts[i][j]/counts[i][0]*1000
counts
#%%
sumc=[]
for j in range(1,len(counts[0]),1):
sumc.append(sum(counts[:,j]))
sumc
for i in range(len(counts)):
for j in range(1,len(counts[0]),1):
counts[i][j]=counts[i][j]/sumc[j-1]*1e6
counts
np.savetxt("tpmonly.csv",counts,fmt="%.6f",delimiter=",")
#%.4f不行,列和不为1000000
- 一些知识点
[序列拼接] 双端测序,原理 + 拼接 (Pandaseq)
双端测序中read1和read2的关系

去除低质量和接头序列?
离线算法—将参考基因组做成索引
基因组Mapping系统索引构建原理
RPKM、FPKM、TPM详解
注:
最后的count文件可能因为某一行太大而在excel中有缺失值,可以在emeditor中设置

在此之后,用,替换\t
或者用以下python代码处理:
#删除counts文件中某些列
def del_counts():
inf = open('data/protein_counts.txt', 'r')
outf = open('data/protein_counts.csv', 'w')
for line in inf:
if line.startswith('#')==0:
list=line.split('\t')
list[1]=list[1].split(';')[0] #染色体只要一个不重复
del list[2:5]
_str = ','.join(list)
outf.write(_str)
else:
outf.write(line)
outf.close()
inf.close()















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









