









现在使用的查看bam文件的方式主要还是需要把bam文件下载到本地,导致下载花费时间较长,偶然间看到igv.js版本,可以把igv部署在web端访问,网上的资料有限,现总结如下,希望可以帮到有同样需求的人。
下载安装
###下载git文件
git clone git@github.com:igvteam/igv-webapp.git
####下载之后就是一个文件夹
cd ./igv-webapp
npm install
npm run build
###上面这两步需要先安装npm才能用,而且非常容易通不过
###安装好了之后会在目录下新生成一个node_modules和dist文件夹
#这一步之后你需要把整个目录放到你的apache的host目录下,如果你是用的apache的话
#######配置调试
#IGV browser默认提供的hg19的参考基因组信息,可以通过igvwebConfig.js配置文件进行更改
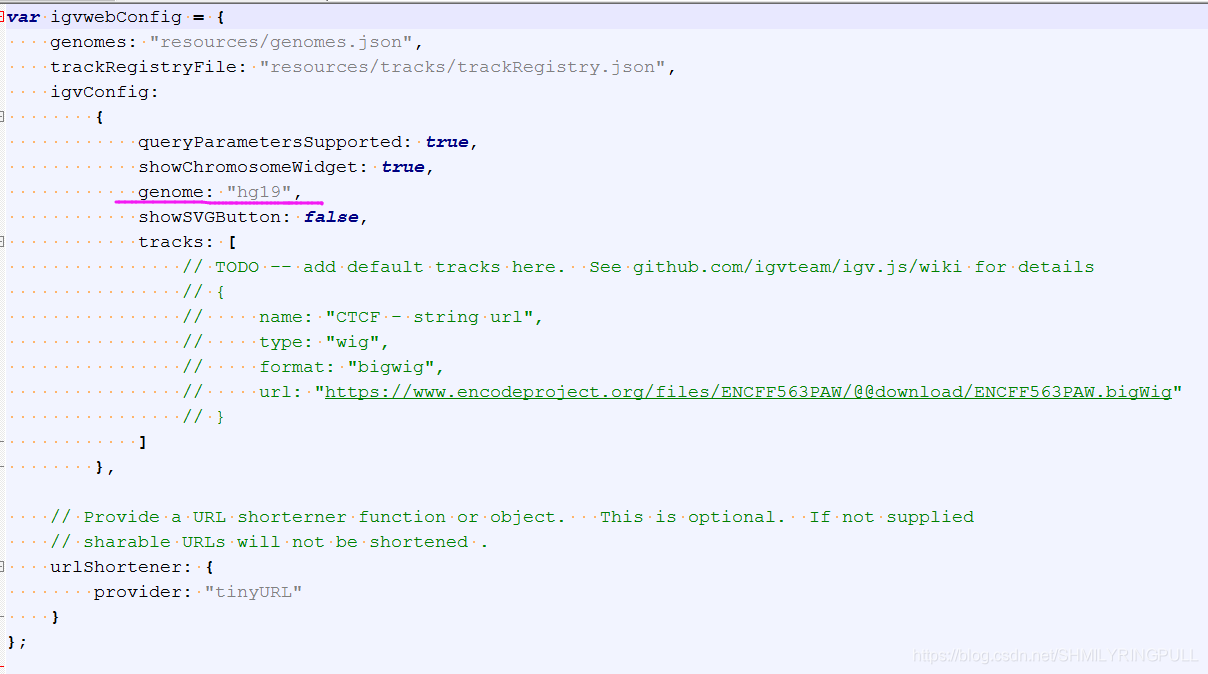
配置个人专属基因组数据
IGV browser的基因组数据是通过resources文件下的genomes.json文件进行读入访问的,可视化的track数据信息存放在resources文件下的tracks文件夹中,通过trackRegistry.json文件进行读入访问,默认会从中读取数据展示,访问数据的加载速度可能会比较慢。 查看genomes.json文件
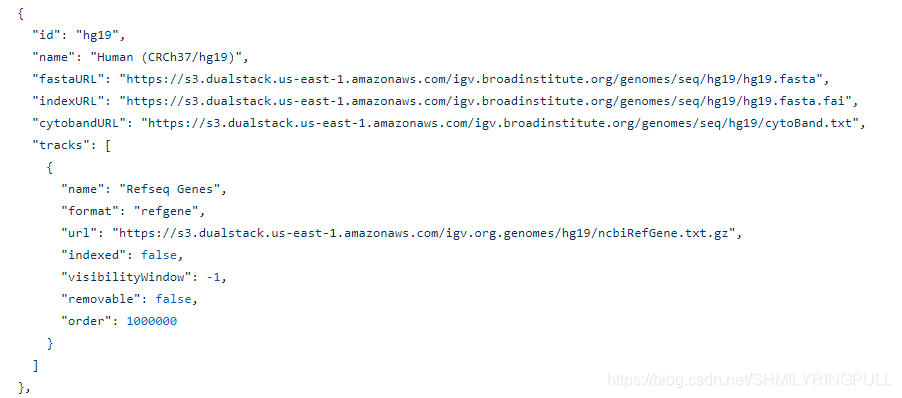
下载并存放个人的基因组数据到data文件中,更改resources文件中的genomes.json文件
cd /hzjn/website/webIGV/data
# 创建个人参考基因组文件夹
mkdir hg19 && cd hg19
# 下载参考基因组相关文件
wget https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/hg19.fasta
wget https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/hg19.fasta.fai
##这两个下载太慢了,我直接把本地常用的拷贝过去使用的命名为:hg19.fa和hg19.fai
wget https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/cytoBand.txt
wget https://s3.amazonaws.com/igv.org.genomes/hg19/refGene.sorted.txt.gz
wget https://s3.amazonaws.com/igv.org.genomes/hg19/refGene.sorted.txt.gz
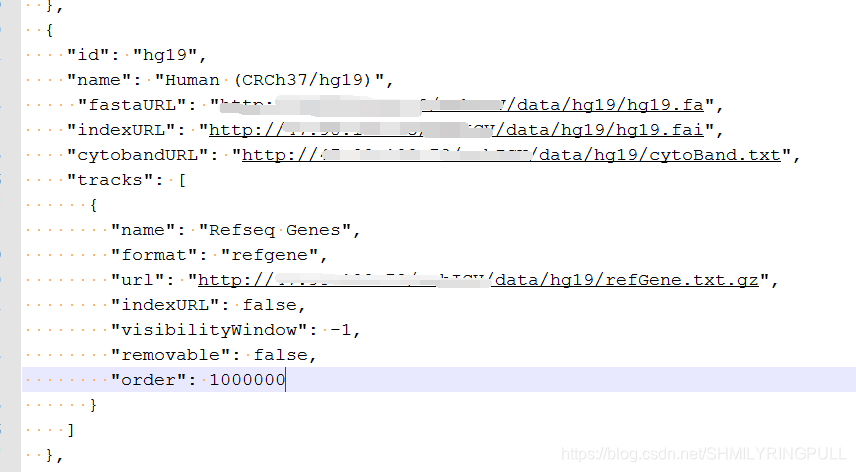
# 更改genomes.json文件
cd ../../resources
vim genomes.json将URL后的地址改为自己的服务器IP端口和数据存放的地址
浏览器中打开网址访问的时候会发现refGene.txt.gz的加载速度特别慢,很明显是从远程现下载,而其他的都是现实from disk cache。
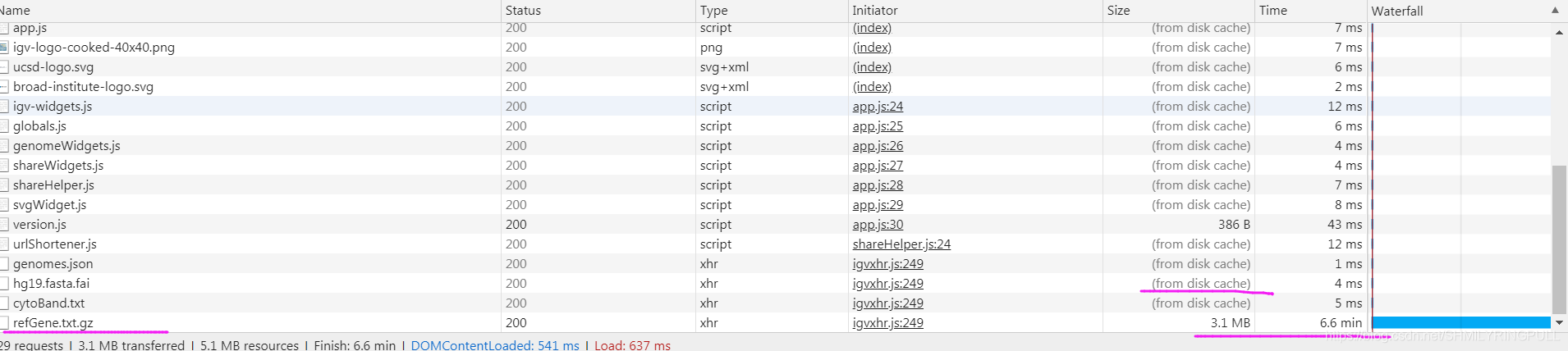
上图显示的是文件名称是refGene.txt.gz,而实际上我们刚才下载的文件名称是refGene.sorted.txt.gz,我试着把refGene.sorted.txt.gz重命名为refGene.txt.gz也不能解决这个问题,应该是网页加载的时候会去判断文件大小,尝试下载这个文件refGene.txt.gz文件
wget https://s3.amazonaws.com/igv.org.genomes/hg19/refGene.txt.gz 的确是有这个文件的
重新下载refGene.txt.gz之后还是不行,chrome浏览器每次都要自动下载,但是firefox浏览器可以加载本地文件成功。哎,这个配置文件也是巨坑啊!
加载调试
这时候在chrome浏览器中输入IP地址以及路径,看是否会加载成功,F12打开控制台查看加载过程,会发现很多都是connect Error,我把index.html中标红的几个都注释掉了,dropbox、google 和twitter都是禁了的,肯定没法访问。注意注释了之后dropbox下拉框就没法用。

https://code.jquery.com/jquery-3.5.1.slim.min.js 这个也是没法访问的,我换成了https://ajax.aspnetcdn.com/ajax/jquery/jquery-3.5.1.min.js 路径文件
我想上面这几个文件如果能下载下来放到本地应该是最好的办法。
到这里再去加载就成功了,导入bam文件的时候,需要把bai文件命名为sample.bam.bai文件才能成功。
track文件我没有配置,大部分情况下使用的人都不会每次去更改配置文件中写入bam文件的。
最终感觉就是这个web版其实对国内用户并不友好,因为很多javascript都被禁了,导致无法正常使用。但是总算可以使用。

















 1481
1481

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









