







官方指南:
Starters guide
Starters guide (workshop version)
其他教程:
teamtomo.org
Youtube:
Dynamo package for cryoET - Daniel Castaño-Diez - I2PC Seminar Series in electron tomography 2021 - YouTube
Session 1. Basics of sub-volume averaging with Dynamo - YouTube
Starters guide (workshop version)
1 Manual particle picking and Dynamo catalogues
Dynamo catalogues 是管理 tomograms 的数据库,并将层析图数据与提取的颗粒联系起来。
通过输入以下命令启动目录管理器:
dcm
在 catalogue manager 打开后,我们以以下方式创建3个 synthetic tomograms,其中包括我们的粒子(热体 thermosomes):
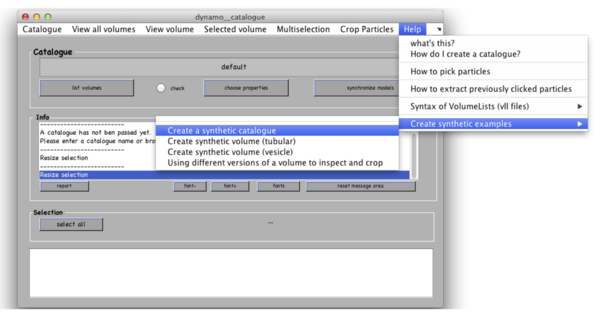
您可以在 catalogue manager 底部的一个表中看到 tomograms 及其元数据的列表。
在处理自己的项目时,您可以通过 Catalogue -> Browse for new volume 将断层图添加到目录中。tomograms 位于目录 testcatalog 中。它们包含少量的噪声和一个与y轴旋转有关的缺失楔。
我们在名为 testcataloge_withmodels 的附加目录中添加了关于 tomograms 中粒子位置的信息。如果需要,可以使用Catalogue -> look for local catalogues在目录管理器窗口中查找。
1.1 Viewing tomograms
选择列表中的第一张层析图,进入View volume>Full tomography file in tomoslice。
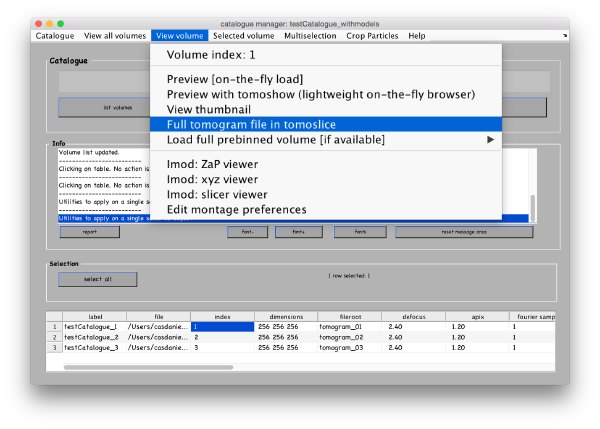
卷浏览器 tomoslicer 将整个 tomograms 加载到内存中,并允许对感兴趣的区域进行注释。
本教程中的层析图很小,您可以将它们直接加载到内存中。对于现实生活中的断层图,请记住,在将文件加载到内存之前,您需要预先保存这些文件。
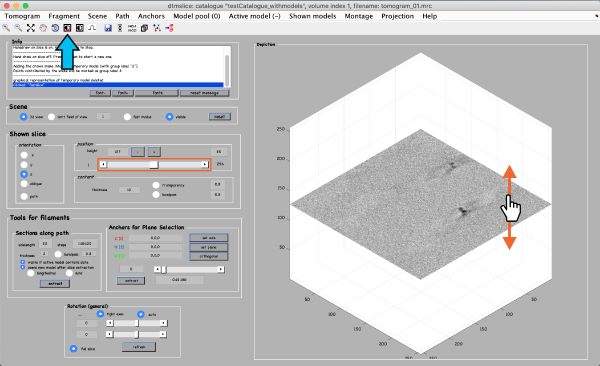
Tomoslice有一组简单的控件,适用于需要通过断层扫描进行斜切片的可视化任务。它使用与其他Dynamo浏览器相同的工具来跟踪您的注释:a pool of models。您可能需要调整对比度(蓝色箭头)。要在层析图切片中移动,您可以使用鼠标滚轮,上下单击并拖动层析图切片(橙色箭头),或者左右移动位置控件(橙色框)。
1.2 Picking and extracting particles in tomograms
在断层图中选取和提取粒子
拾取粒子的坐标由称为模型的数据类型表示。在建模器窗口( tomoslicer window)中,转到Model Pool -> Create new model in pool (choose type) -> General。这是最简单的模型类型,其中每个 clicked/model point 对应于单个孤立的粒子。
现在你可以上下浏览断层图,把鼠标放在粒子的中心,按键盘上的[c]键来添加一个新的模型点。(注意Help -> all hot keys选项列出了不同按键的不同操作)。退格键删除最后单击的点(您也可以使用右键单击删除单个点)。

当你完成了大约10个粒子点击活动 Active model > Update Crop points在 tomoslice window
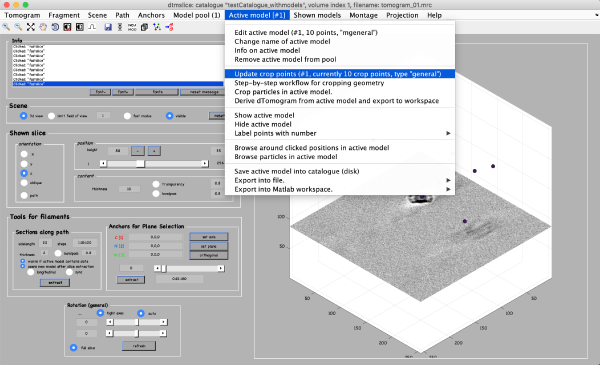
通过Active model -> Save Active model into catalog(disk)将模型保存到目录中,并关闭 slicer window。
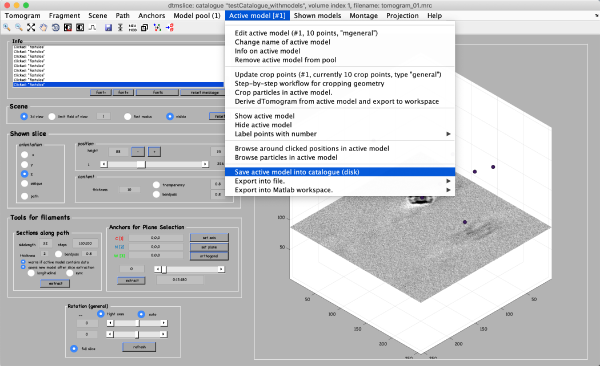
为 tomograms 2和3选择粒子。总的来说,在生成的数据集中大约有30-40个粒子。
当您打开一个新的断层图时,请确保在被要求时从内存中删除模型池。这对存储在磁盘中的模型没有影响,并且为了确保您没有混合来自不同层析图的模型,这是必要的。
1.3 An alternate visualization: orthogonal projections
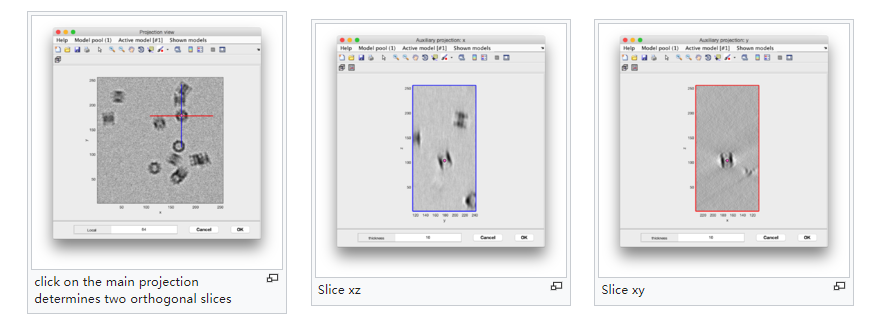
点击菜单上的Projection -> project full shown fragment along z (仍然在dtmslice),你会得到一个屏幕,其中x-y投影的 tomogram 显示。
使用二次点击来启动横过该点的x-z和y-z平面的正交视图。这些视图也可以用来点击粒子,以防标准视图不够用。
2 Extracting particles from tomograms
在目录管理器窗口中,选择要从中提取粒子的断层图行。您可以使用鼠标单击并按住[ctrl]键逐个选择它们,或者单击Select all 按钮。然后,转到Crop Particles -> Open Volume List Manager。
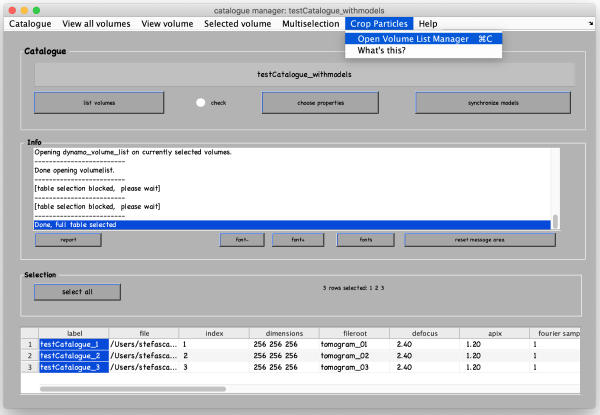
将打开一个新窗口,底部列出目录中的所有型号。选择(通过复选框)您刚刚单击的general类型的模型。点击Create list ,然后Crop particle。
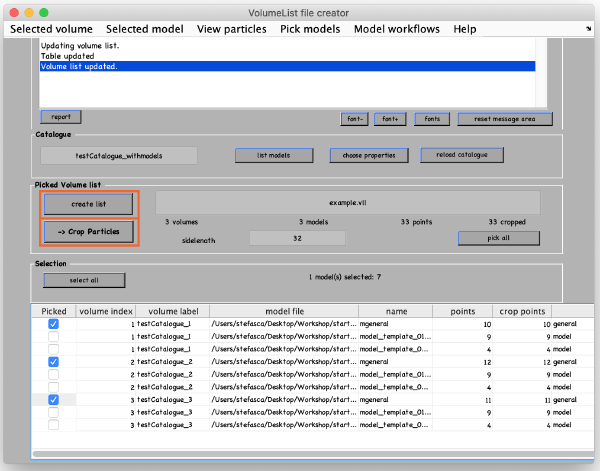
在新窗口中,将数据名称更改为有意义的名称,例如thermosomeParticles,将边长从32更改为48,然后单击start cropping。
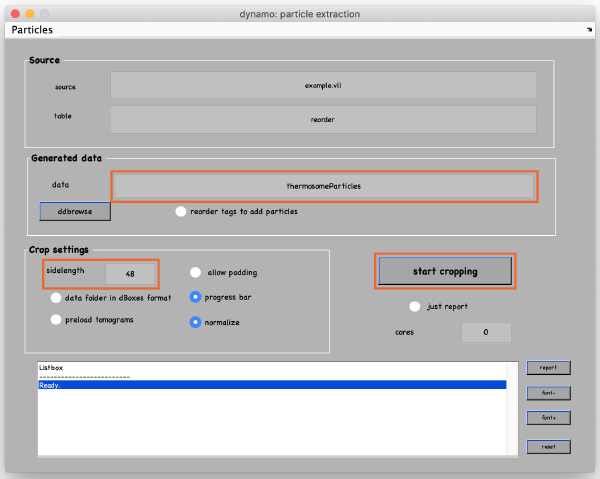
裁剪完成后,通过点击ddbrowse(在上面的窗口中)来浏览裁剪后的粒子。打开一个新窗口,您可以简单地单击show。
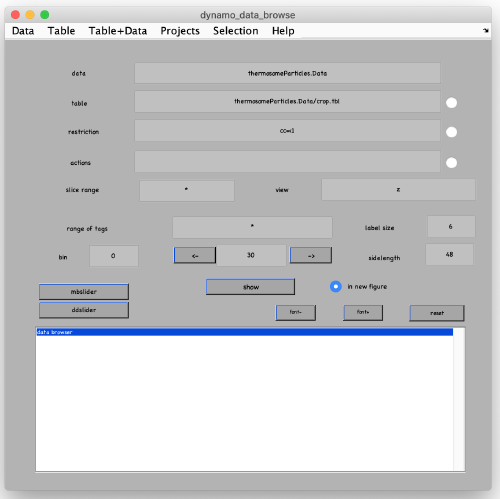
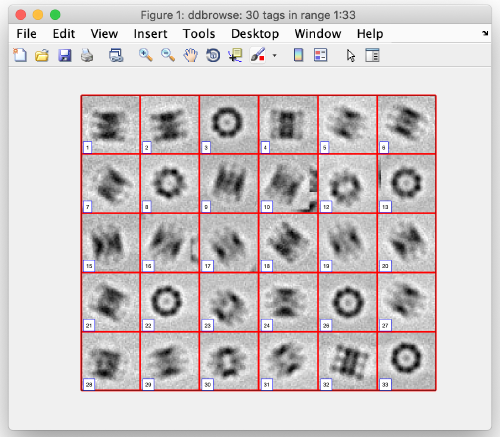
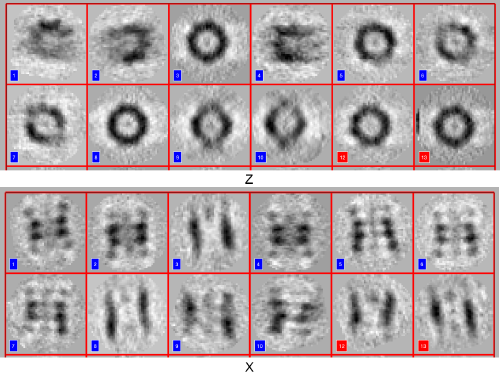
您可以看到所有裁剪的3D subtomograms的2D表示(投影)。确保你的粒子是很好的中心和 fit the box。
我们还引入了dynamo table的概念:关于每个粒子的信息存储在表中。每个粒子在表中都有一个条目,包含移动和旋转来描述它的方向(默认情况下,这些值初始化为零)。它还包含粒子ID(标签),missing wedge 的方向和其他。要获得完整的信息,请键入命令dthelp。Dynamo目录在粒子提取过程中生成一个初始表。 Its location is in particles/crop.tbl.
3 Subtomogram alignment and averaging
3.1 Initial reference generation
我们希望将提取的层析图对齐到一个公共参考。
为此,我们需要一个 initial reference (稍后将在迭代校准期间进行细化)。在这里,我们通过手动对齐一些粒子并平均它们来创建这样一个初始参考。我们使用命令dgallery为一些将用于生成初始参考的粒子生成初始方向。关闭所有打开的Dynamo窗口,并在Dynamo命令行中输入:
dgallery('data','thermosomeParticles.Data');
点击load加载内存中的所有粒子,移动shown bar 显示粒子,使用x-, y-和z-按钮可以看到不同的视图:
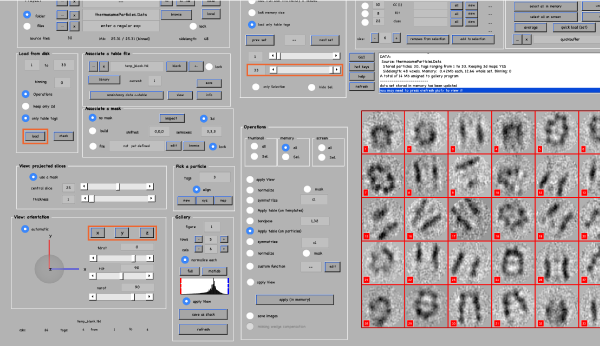
要手动对齐粒子,请对大约10个粒子执行以下操作:
- 将鼠标放在粒子中心并按下
[c]键(这将使粒子居中)。 - 将鼠标放在其顶部(或底部)部分并按下
[n]键(这将对齐粒子)。 - 点击粒子,确保它的数字从红色变为蓝色(蓝色表示它被选中进行进一步处理)。要取消选择粒子,请使用右键单击。
如果需要,在x, y和z视图之间更改以纠正方向。这样做只是为了创建一个初始引用,所以方向不需要非常精确。
通过单击右上方的quick save按钮保存选中的标签和相应的表。它保存quickbuffer.tbl 和 quickbuffer.tags。标记到稍后将使用的硬盘驱动器。为了生成平均值,你需要在粒子上应用表格。为此,单击粒子选择字段中的average按钮。
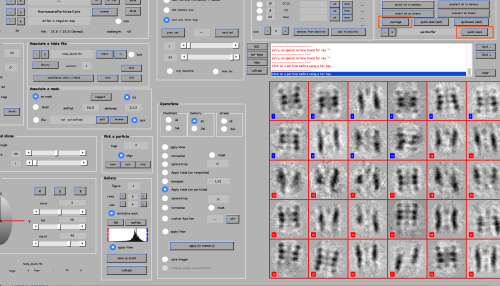
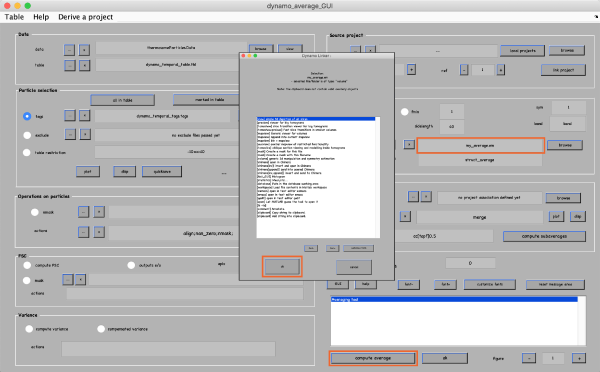
它会打开一个带有许多控件的新窗口。单击窗口底部的compute average。然后,右键单击输出文件名并在下一个窗口中单击ok以查看结果。如果你对结果不满意,关闭窗口并改进你的手动对齐和/或添加更多的粒子到平均值。

3.2 Alignment projects
subtomograms 与其平均值的迭代对齐是由可以在各种高性能计算环境中运行的 dynamo 项目执行的。要运行对齐项目,您需要粒子、初始参考和表。所有这些文件都是我们之前生成的。通过键入启动对齐项目GUI:
dcp
在新窗口中,执行以下操作:
- 添加项目名称
drun1,按enter键并在弹出窗口中选择create a new project。 - 点击粒子并提供粒子文件夹名称
thermosomeParticles.Data,单击ok。 - 单击表格并提供表格名称
thermosomeParticles.Data/crop.tabl,单击ok。 - 单击模板并提供初始引用名称my_average.em。单击ok。
- 点击蒙版,只需点击
use default masks。单击ok。你也可以指定椭球蒙版的半轴,或者去Mask editor制作更复杂的蒙版。注意Dynamo默认使用Rossman相关,它消除了与硬(非软)掩码相关的工件。
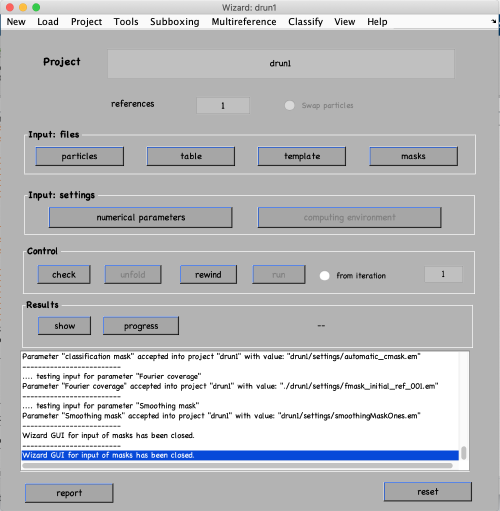
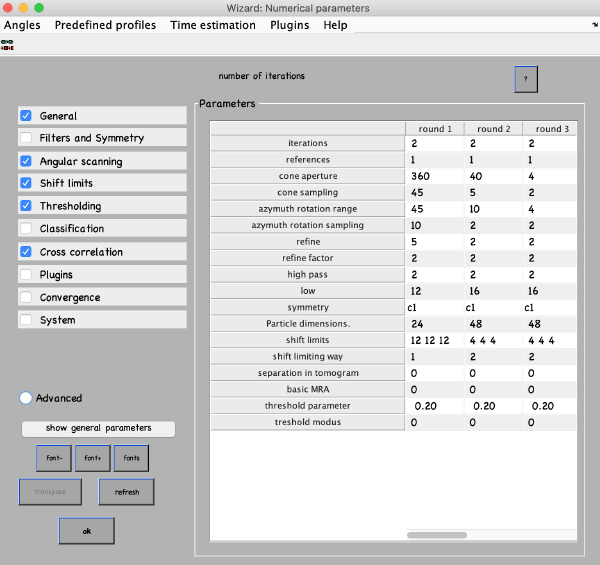
点击数值参数numerical parameters来设置关于校准项目的不同迭代的所有参数和细节。您可以用鼠标选择表中的任何参数,然后单击?参数窗口右上方的按钮,以查看参数的说明。需要考虑的最重要参数是:
- Number of iterations:进行3轮,每轮2次迭代。第一轮将是一个具有粗糙角度步骤的全局搜索,接下来的几轮将用于细化。
- Angular search ranges:圆锥孔径是表中定义的初始方向周围的前两个欧拉角的扫描范围。360度是整个扫描范围。方位旋转范围定义了围绕粒子的新垂直轴的旋转范围。
- High- and lowpass values:傅里叶体素来限制使用的频率范围。
粒子尺寸:定义为子体积的边长。如果你设置一个较低的值,粒子将在特定的回合中被下采样。这将加快这一进程。 - Refine:在每次角度扫描后,搜索步骤被细化因子减少。这是重复的精炼次数。也就是说,如果你的锥体采样是10度,细化是3,细化因子是2,那么10度,5度,2.5度和1.25度将被采样。这是角搜索空间的优化。
- Shift limits:限制粒子从框中心移动(如果移动限制方式为1)或从先前估计的中心移动(如果移动限制方式为2)。先前对移动和旋转的估计来自输入表,并在每次迭代结束时更新。
- Symmetry:如果你知道你的蛋白质的对称性,它将加快收敛,并得出更高的分辨率。
按如下参数配置,单击“ok”。
单击Computing environment并选择您的计算环境(如果您在研讨会期间使用独立版本,请选择standalone)。设置CPU核数和并行化平均步长到最大可用(例如 24)。其余部分保持原样,然后单击ok。在对齐GUI中单击check并unfold。要运行项目:
- 在Matlab会话中,您只需点击“
run”即可。 - 在一个独立的项目中,打开一个新的终端,加载Dynamo(但不运行它)并通过键入
./drun1.exe来运行项目更方便。(博主建议使用此方法和下面的命令来运行和查看)
当项目运行时,打开另一个Matlab窗口(或在独立打开另一个运行Dynamo的终端),您可以通过输入来监控对齐项目的进度:
dvstatus drun1
当项目运行时,您可以通过输入以下命令查看中间结果的投影:
ddb drun1:a:ite=* -j c10
或者输入最新的平均值:
ddb drun1:a -v
最终结果应该类似于:

4 Subtomogram classification
为了演示分类示例,我们提供两种大小稍有不同的颗粒。然后我们可以用简单的多参考分析(Multi-reference Analysis,MRA)对它们进行分类。要做到这一点,我们首先生成一个有2个粒子群的数据集。为此,输入:
dynamo_tutorial('data_classification','M',8,'N',8);
它生成两组粒子,每组8个粒子,包括它们的表。
MRA背后的思想如下:
粒子与几个参考对齐(这里只有2个)。每次迭代后,每个粒子只分配给一个最适合它的参考。这个过程是反复重复的。相似的粒子与相似的参考有更高的相关性,最终会聚集在一起。为了将其分类为N个类,我们需要N个初始参考和N个初始相同的表。按照以下步骤运行MRA:
- 通过在 alignment GUI的项目名称中输入“drun2”来创建一个新项目。
- 将引用数设置为2。
- 激活
Swap particles按钮。 - 转到
particles并在particle data中添加data_classification/data。单击ok。 - 转到
table并在clone this中输入data_classification/real.tbl进入并按下copy。单击ok。 - 进入
template,clone this file →data_classification/original_template.em加上一些额外的噪声,振幅为1。 - 点击
masks,点击use default masks。单击ok。 - 单击
numerical parameters,设置以下参数。单击ok。
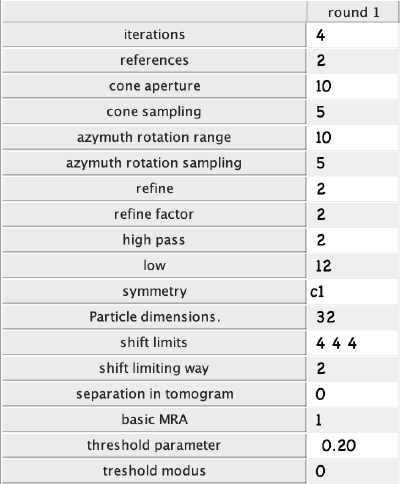
选择与前面对齐项目中相同的计算环境,并以相同的方式运行项目。MRA项目通常运行时间更长,因为每个粒子将与两个引用对齐。然而,在这里,我们以一种过程仍然快速的方式选择参数(例如,小维度)。与之前一样监控项目进度:
dvstatus drun2
并使用命令可视化结果:
ddb drun2:a:ref=* -j c5
你应该得到两个平均值,其中一个类的粒子略大于另一个类。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









