







我们都知道进行细胞间通讯分析最常用的两个软件是CellChat和CellphoneDB,大多数常规的通讯分析可以得到不错的效果,不过常常也会遇到一些想要了解的通路和配受体找不到的情况,这个时候就需要更加个性化的数据库和方法的支持。
根据2023年NC发表的一篇文献,
NeuronChat是专门为推断神经细胞之间的通讯而设计的,它考虑了神经元特有的长距离轴突和树突连接以及通过神经递质信号进行通讯的特点,而传统方法多基于短程旁分泌/自分泌信号,主要通过配体扩散或细胞物理接触进行通讯,不适用于神经元之间的通讯。
NeuronChat开发了一个手动整理的神经信号分子相互作用数据库NeuronChatDB,包含373个条目,涵盖人类和小鼠的神经细胞间分子相互作用,包括小分子神经递质、神经肽、缝隙连接蛋白、气体信号分子和突触粘附分子等。
于是我们找到了下载路径开始实践https://github.com/Wei-BioMath/NeuronChat
环境导入
首先导入R环境和需要的包
library(NeuronChat)
library(Seurat)
library(CellChat)
library(patchwork)
library(dplyr)
library(ggalluvial)数据导入
导入需要的标准化后的矩阵和注释文件
data <- readRDS("your.rds")
#数据需要经过标准化处理
#data <- NormalizeData(data, verbose = TRUE)
log_data <- as.matrix(data@assays$RNA@data)
meta <- data@meta.data注意确保需要进行通讯分析的注释标签在@meta.data的某一列中,标准化后的矩阵存放在data@assays$RNA@data中
映射分组
还有可选的一步,添加注释标签之间的映射分组,主要是为了后续大类和子类的可视化
df_group <- meta[!duplicated(meta$sub2), c('major', 'sub2')]
group <- structure(df_group$major, names = df_group$sub2)比如我这里的‘major’就是大类标签,‘sub2’是更加精细化的亚类标签
创建NeuronChat对象
x <- createNeuronChat(log_data, DB='human', meta=meta, group.by = meta$sub2)
x <- run_NeuronChat(x,M=100)利用准备好的数据就可以开始计算了,这里我选择的是人的数据库
参数 `M` 用于指定进行置换检验(permutation test)的次数。置换检验是一种统计方法,用于评估细胞间通讯网络中各个连接的显著性。具体来说,通过随机打乱细胞的分组标签,然后重新计算每个置换后的通讯强度,从而得到一个经验分布。通过这个分布,可以计算出原始通讯强度的 p 值,从而判断该通讯连接是否显著。
在 `NeuronChat` 的默认设置中,`M` 的值通常设置为 100。虽然增加 `M` 的值可以提高 p 值的准确性,但也会增加计算时间。根据研究,使用 100 次置换和 1000 次置换得到的 p 值结果具有很好的一致性,因此在大多数情况下,使用默认的 100 次置换可以在保持准确性的同时节省计算时间。
可视化展示
这里的可视化和CellChat类似(本身就嵌入了),使用自带的功能就比较美观了
权重网络图
合并各通路权重绘制网络图,vertex.label.cex可以调节标签字体大小
net_aggregated_x <- net_aggregation(x@net,method = 'weight')
netVisual_circle_neuron(net_aggregated_x)
#netVisual_circle_neuron(net_aggregated_x, vertex.label.cex = 0.8)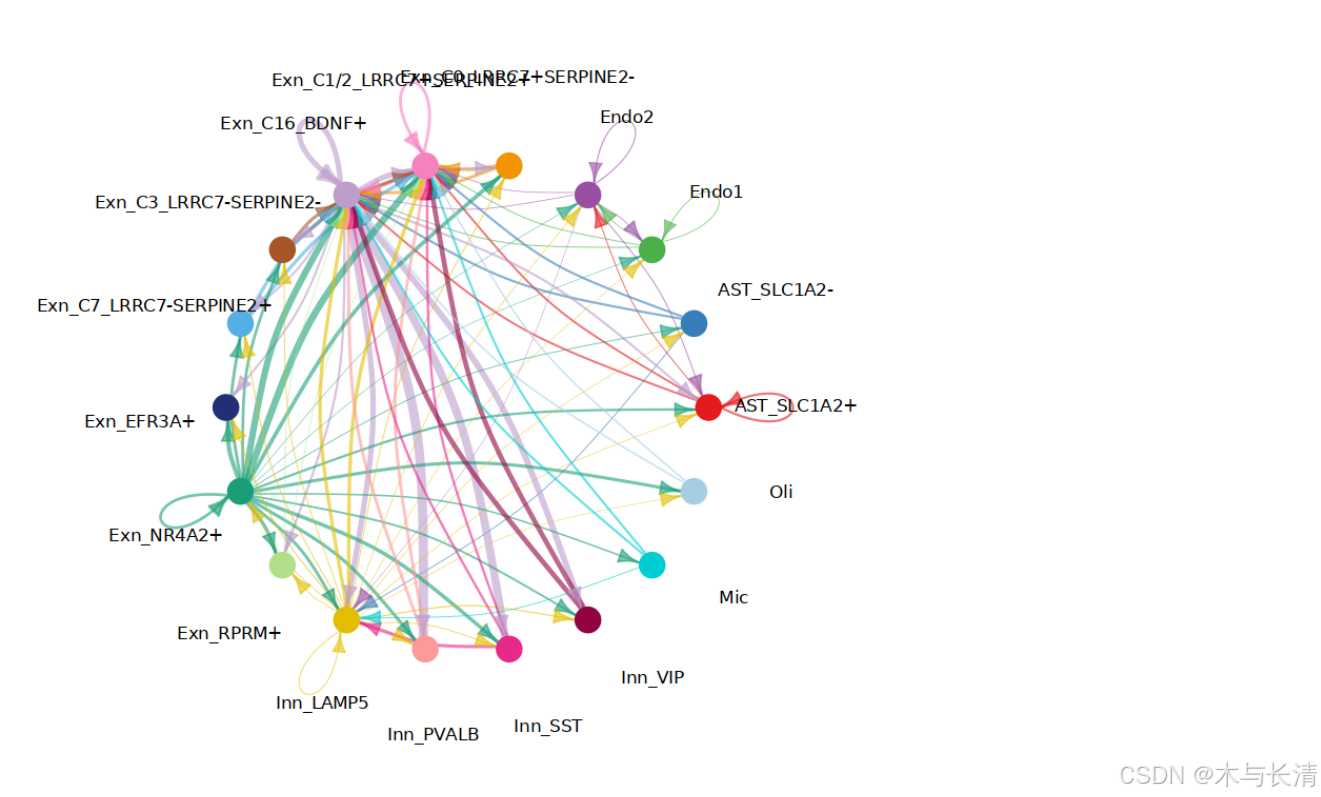
权重网络表格
为了更详细检查每一个网络情况,建议输出表格保存到本地,所有网络存放在x@net
net_data <- x@net
net_weights <- sapply(net_data, function(mat) sum(mat, na.rm = TRUE))
sorted_net_weights <- sort(net_weights, decreasing = TRUE)
net_weight_table <- data.frame(Network = names(sorted_net_weights), Weight = sorted_net_weights)
write.csv(net_weight_table, file = "network_weight_sort.csv", row.names = FALSE)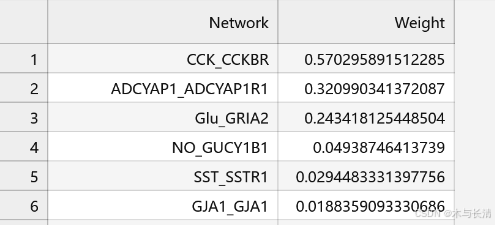
热图
如果有感兴趣的具体配受体通路,可以查找名称后绘制热图,names(x@net)可以列出所有名称
heatmap_single(x,interaction_name='ADCYAP1_ADCYAP1R1',group=group)这里的group就是准备数据是的映射分组,可视化中就可以按大类分布
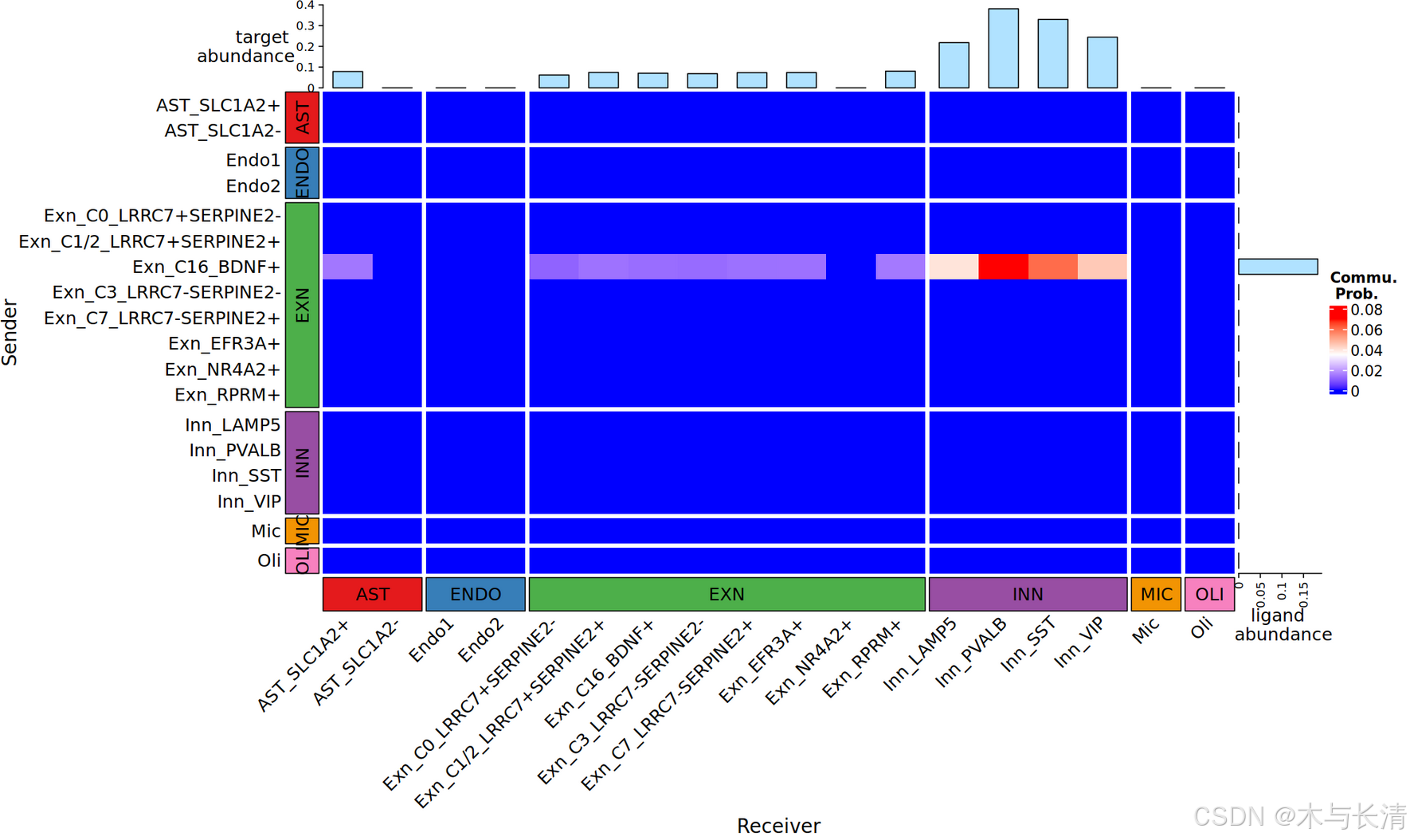
配受体提琴图
如果想要可视化配受体基因的贡献小提琴图,可以使用
lig_tar_heatmap(x,interaction_name='ADCYAP1_ADCYAP1R1',width.vector=c(0.38,0.35,0.27))width.vector是子图间隔大小
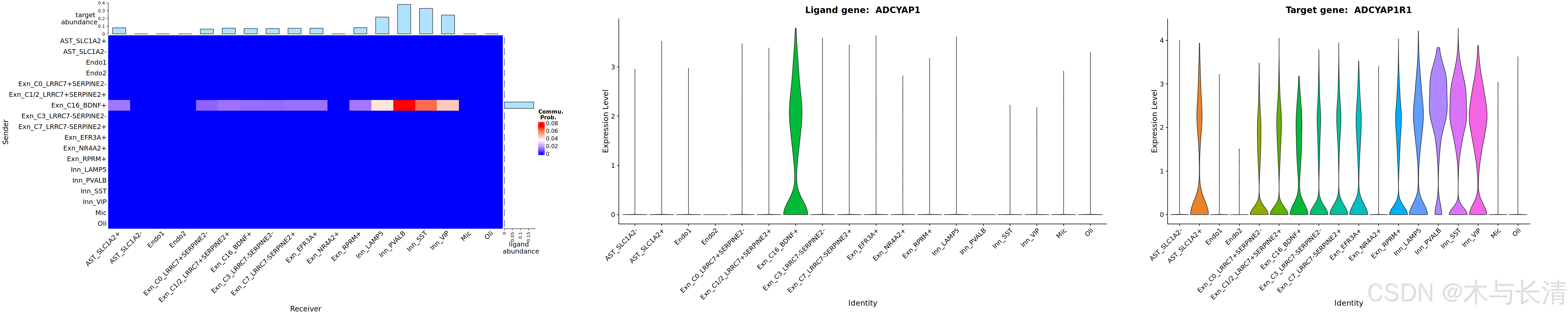
非零通路打分表
输出包含上述传入传出的所有非零通路表格
net_data <- x@net
net_weights <- sapply(net_data, function(mat) sum(mat, na.rm = TRUE))
sorted_net_weights <- sort(net_weights, decreasing = TRUE)
# 筛选出非零的interaction
non_zero_interactions <- names(sorted_net_weights[sorted_net_weights > 0])
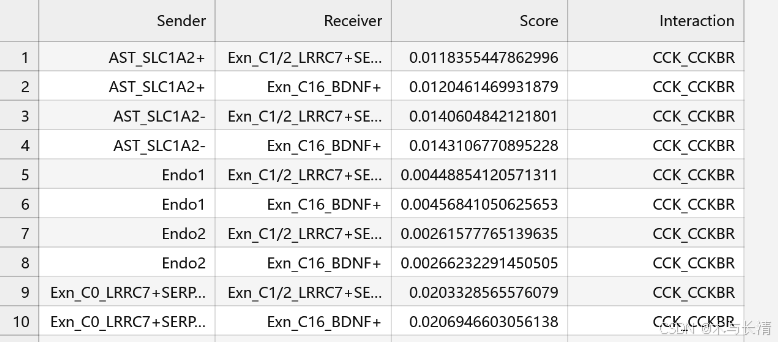
all_communication_data <- data.frame(Sender = character(), Receiver = character(), Score = numeric(), Interaction = character())
for (interaction in non_zero_interactions) {
p <- heatmap_single(x, interaction_name = interaction, group = group)
score_matrix <- p@matrix
senders <- rownames(score_matrix)
receivers <- colnames(score_matrix)
for (i in 1:nrow(score_matrix)) {
for (j in 1:ncol(score_matrix)) {
if (score_matrix[i, j] != 0) {
new_row <- data.frame(Sender = senders[i], Receiver = receivers[j], Score = score_matrix[i, j], Interaction = interaction)
all_communication_data <- rbind(all_communication_data, new_row)
}
}
}
}
write.csv(all_communication_data, "all_communication_scores.csv", row.names = FALSE)效果如下
还有一些不太常用的可视化,可以按需绘制
传入传出模式图
x<- identifyCommunicationPatterns_Neuron(x, slot.name = "net", pattern = c("outgoing"), k=4,height = 18)
x<- identifyCommunicationPatterns_Neuron(x, slot.name = "net", pattern = c("incoming"), k=4,height = 18)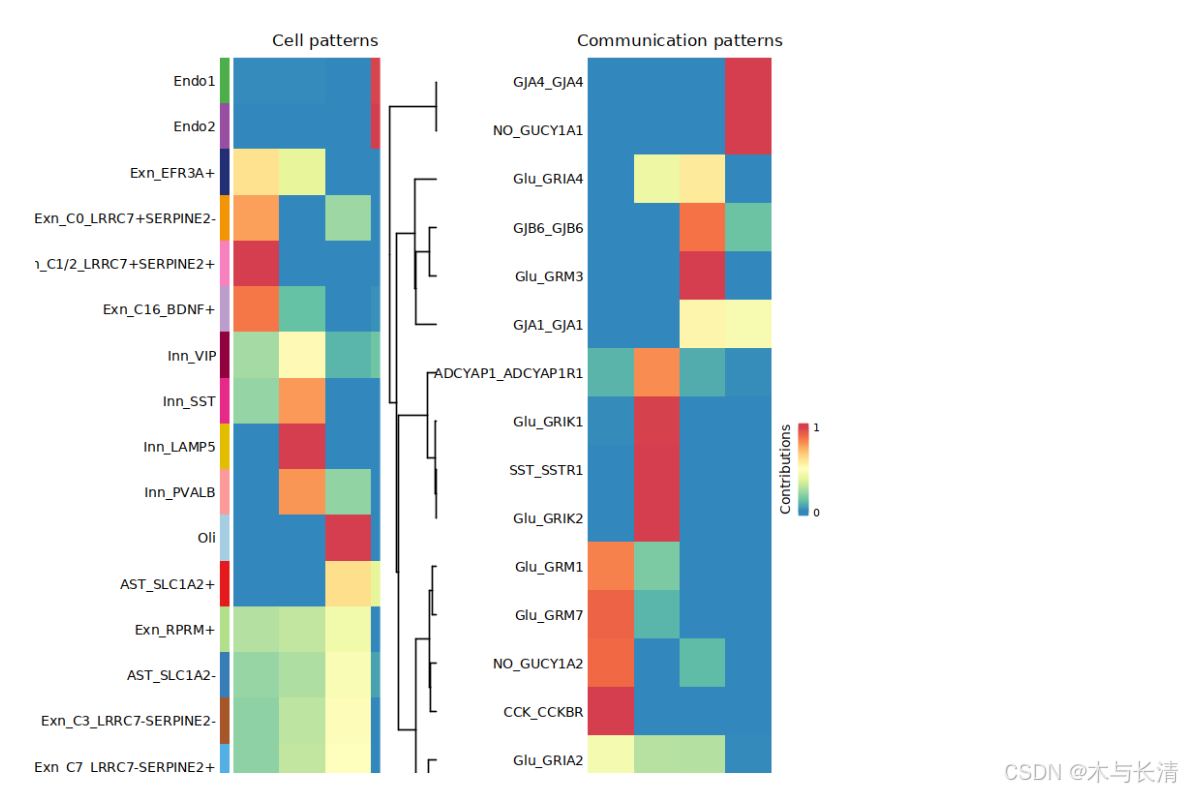
netAnalysis_river_Neuron(x,slot.name = "net", pattern = c("outgoing"),font.size = 2.5,cutoff.1 = 0.5,cutoff.2=0.5)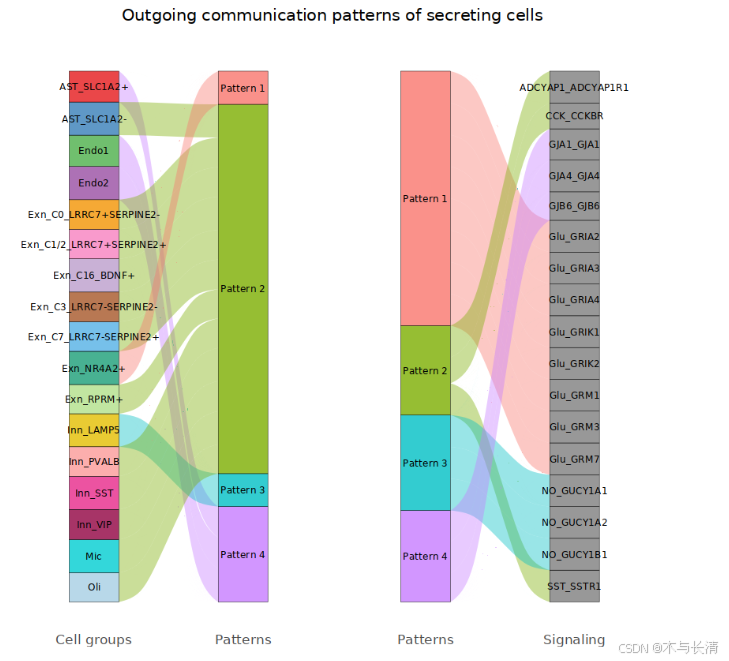
相似性分类图
x <- computeNetSimilarity_Neuron(x,type='functional')
x <- CellChat::netEmbedding(x, slot.name = "net_analysis", type = "functional")
x <- CellChat::netClustering(x, type='functional',slot.name = "net_analysis",k=5)
netVisual_embedding_Neuron(x, type = "functional", label.size = 5,pathway.remove.show = F)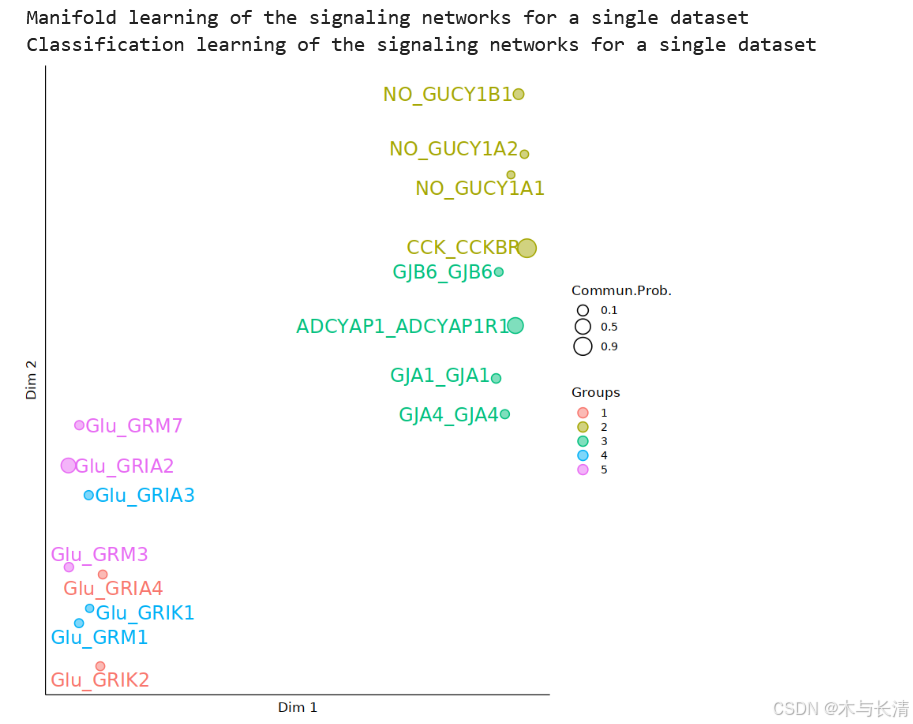
netVisual_embeddingZoomIn_Neuron(x, type = "functional", nCol = 2,label.size = 3)

















 2801
2801

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









