







示例数据来自2019.10的NC,其单细胞数据被上传到GEO网站
常见的单细胞数据应包括如下三个文件,带有单细胞计数的matrix矩阵文件,基因名称的列表文件,以及包含样本注释的metadata文件。文件格式一般为mtx、txt、tsv或者其压缩格式。
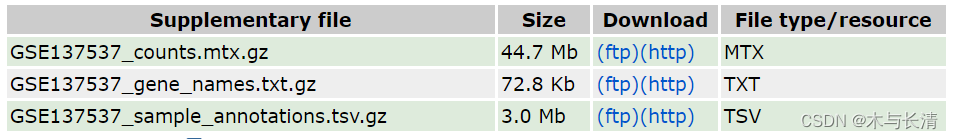
scanpy包是目前比较通用的单细胞数据分析的软件包,其数据存储格式也较为清晰,这里我们尝试将单细胞的三个文件转换成适用于scanpy分析的anndata对象。
首先导入相应的包,下载好数据文件并提前指定路径
import scanpy as sc
import os
import math
import itertools
import warnings
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
mtx_file = '/data/work/scRNA_load/2019.10.NC/GSE137537_counts.mtx.gz' # MTX格式文件路径
genes_file = '/data/work/scRNA_load/2019.10.NC/GSE137537_gene_names.txt' # 基因名称文件路径
annotation_file = '/data/work/scRNA_load/2019.10.NC/GSE137537_sample_annotations.tsv'
将单细胞计数文件直接导入成anndata,查看数据
adata = sc.read_mtx(mtx_file)
adataAnnData object with n_obs × n_vars = 19719 × 20091
"obs"和"vars"是"AnnData"对象中两个重要的属性,分别代表观测值(Observations)和变量(Variables)。在单细胞RNA测序数据中,每一行代表一个单细胞样本(通常是一个细胞),而每一列则是样本中的一个特征或基因。"obs"属性存储了与每个单细胞样本相关的元信息,比如细胞类型、样本来源、处理条件等。通常,它是一个表格或字典格式,其中每一行对应于一个单细胞样本,每一列表示一个元信息。
而每个样本的每个特征或基因都具有相应的值。"vars"属性存储了与每个特征或基因相关的元信息,比如基因名称、基因类型、基因表达水平等。通常,它是一个表格或字典格式,其中每一行对应于一个特征或基因,每一列表示一个元信息。
为了检查我们的数据结构是否正确,用pandas包导入metadata信息验证
df = pd.read_csv(annotation_file, sep='\t')
df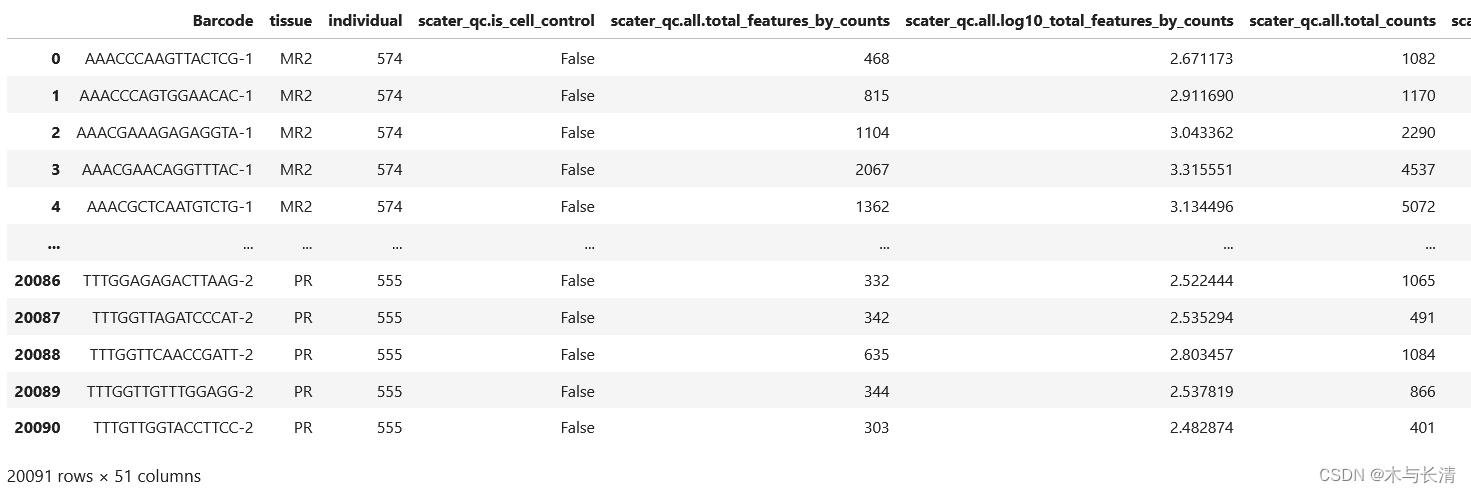
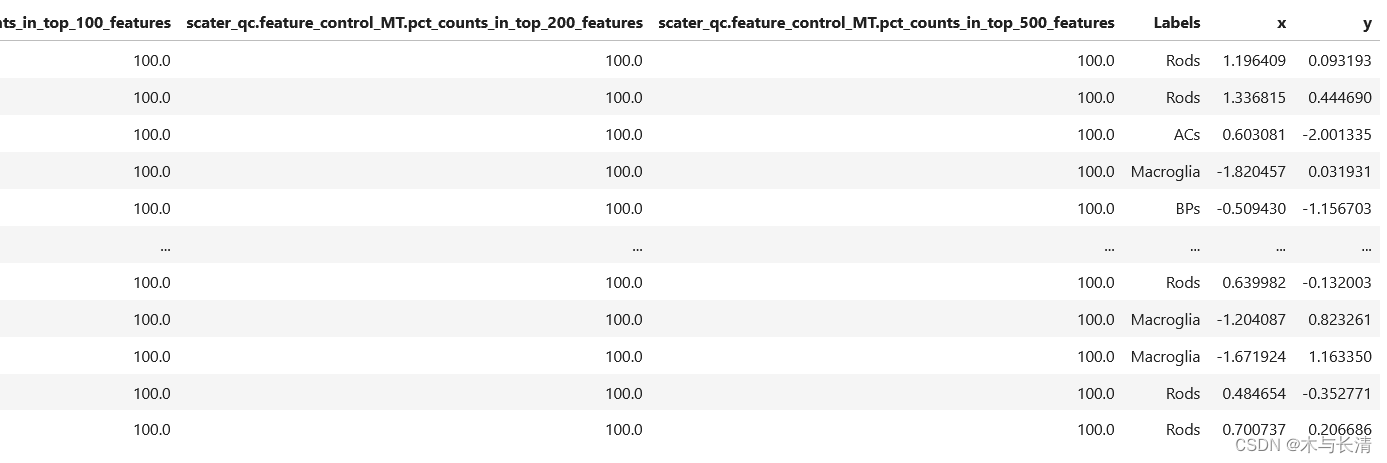
由metadata可知,20091是样本的单细胞数目,应该对应anndata的观测值,因此我们需要转置一下数据结构,即
adata = adata.transpose()
print(adata)AnnData object with n_obs × n_vars = 20091 × 19719
接着可以向变量值中添加基因名称
adata.vars_names = open(genes_file).read().splitlines()最后将基因注释信息存储到.obs的观测列中,原始数据中注释信息在‘Labels’列,我们将其存储为anndata数据的‘celltype’列,同时提取添加两个特征计数列
adata.obs_names=df.index
adata.obs['celltype'] = df['Labels']
adata.obs['nCount_RNA'] = df['scater_qc.all.total_counts']
adata.obs['nFeature_RNA'] = df['scater_qc.all.total_features_by_counts']
# 查看前十行
print(adata.obs.head(10))celltype nCount_RNA nFeature_RNA 0 Rods 1082 468 1 Rods 1170 815 2 ACs 2290 1104 3 Macroglia 4537 2067 4 BPs 5072 1362 5 Macroglia 1205 421 6 Macroglia 4237 2063 7 BPs 1914 896 8 Macroglia 5790 2534 9 Rods 933 721
查看现在的数据结构,可以进行一些规范化处理
sc.pp.normalize_total(adata)AnnData object with n_obs × n_vars = 20091 × 19719
obs: 'celltype', 'nCount_RNA', 'nFeature_RNA'
最后保存为.h5ad格式
output_file = '/data/work/scRNA_load/2019.10.NC/GSE137537.h5ad'
adata.write(output_file)如果需要进行后续分析,直接读入即可
adata = sc.read_h5ad('/data/work/scRNA_load/2019.10.NC/GSE137537.h5ad')
adataAnnData object with n_obs × n_vars = 20091 × 19719
obs: 'celltype', 'nCount_RNA', 'nFeature_RNA'















 1600
1600











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









