







vcf文件做记录个体或群体突变的文件格式,在生物信息学应用中举足轻重。主流的生物信息分析软件,在处理变异信息时,也基本上需要考虑支持解析或输出vcf格式的文件。本文在介绍vcf文件格式的基本格式的同时,对vcf文件记录的细节进行描述。希望对广大开发者和生物信息学从业人员起到帮助。
1.vcf文件概述
vcf文件格式是变异结果存储的标准格式,一般多用于单核苷酸变异(SNV)或小片段的插入缺失(indels)的结果记录。除此之外,vcf文件也可以存储其他变异形式,比如CNV(拷贝数变异)、SV(结构变异)等,但目前难以形成主流。基因组结构类变异,目前相对较多的依然是bed或bedpe文件,后面会陆续为各位进行介绍。
SNV 是基因组上单个位置的替换。|比如,在参考基因组上记录为 A ,通过检测,某个体由于个体差异或突变,在相同位置变异为C。Indel是指插入或缺失,例如在参考基因组上,某位置为ATCCA,在个体基因组上为ACA(中间位置的TC缺失),则记为A--CA,该位置为deletion。同理若个体基因组存在插入(insertion)情况,与deleltion一样也可以进行记录。insertion和deletion合称为indel。
vcf文件主要有三种模式:
- 第一种为仅有位点信息,即对变异发生的位置和变异本身
- 第二种为个体变异记录的是某个个体或个体组织的突变情况
- 第三种为群体变异检测信息,记录的为突变在群体或家系中发生情况。
这三种类型文件虽然在记录内容上有所差别,但是都遵循vcf的基本规则。下图为vcf文件实例:
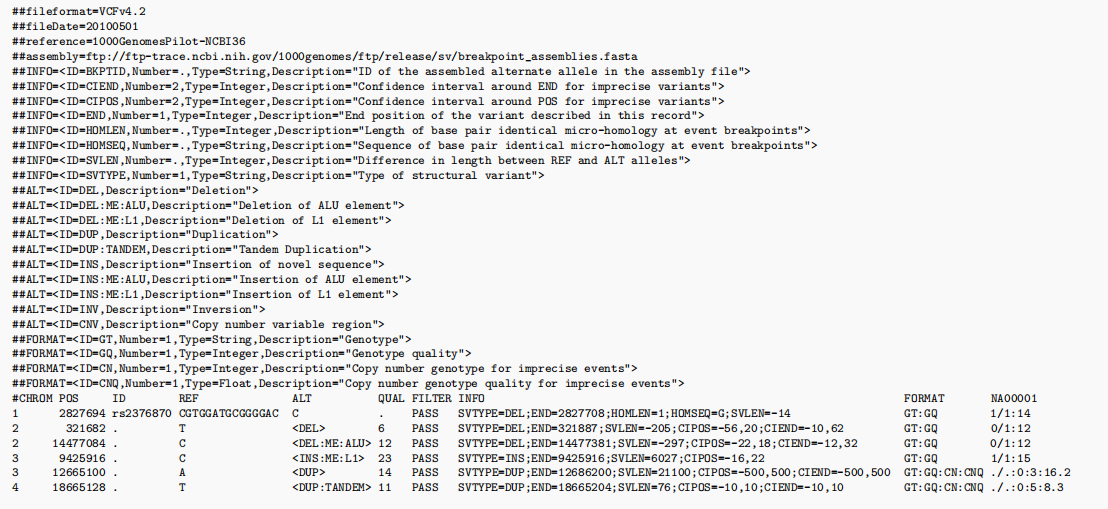
注意: 从上述说明中可以看出,单核苷酸的记录其实相对容易,但是对indel变异而言,由于插入缺失片段的长度不定,其位置并非固
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章















 8834
8834











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









