






GATK4 实用技巧
前言
本篇笔记围绕GATK4流程中注意事项进行介绍,包括如何选择合适的线程和内存?如何按步骤进行数据预处理。
-
GATK实战工作流程 -
脚本优化方法 -
参数设置技巧
GATK 全称“基因组分析工具箱” Genome Analysis Toolkit,它是一组命令行工具,用于分析高通量测序数据,主要侧重于变异发现。这些工具可以单独使用,也可以链接在一起形成完整的工作流程。
所需数据与软件
需要有简单的Linux知识储备和高通量基因组学基础,流程中用到的软件如下:
GATK 4.3.0.0
fastp 0.20.1
bwa 0.7.17
samtools 1.11
mosdepth 0.3.0
示例数据来自ERP001960项目,数据结构为illumina测序原始数据,包含三个样本,如果想重复测试需要大概700GB空间。 
数据预处理
预处理的步骤包括质控、去接头、排序等,需要用到FATSP等工具,为了提高效率建议直接用fastp标准输出到BAM文件,避免不必要的磁盘读写,用管道串联起来能提高工作效率。
无论是否进行排序,都不要直接将未压缩的sam文件写入磁盘!最好用管道直接把两步合为一起生成排序去重之后的bam文件
fastp去除接头
使用fastp程序进行操作,为了提高数据吞吐效率,将默认输出信息定向到/dev/null中,避免磁盘读写。
fastp -i /lscratch/${SLURM_JOB_ID}/$(basename $r1) \
-I /lscratch/${SLURM_JOB_ID}/$(basename $r2) \
--stdout --thread $threads \
-j "${logs}/fastp-${SLURM_JOB_ID}.json" \
-h "${logs}/fastp-${SLURM_JOB_ID}.html" \
> /dev/null
下图展示了不同线程与处理速度的关系,很显然啊!这里如果超过6个线程就没多大意义了,设置6个线程就能达到最佳计算效率。 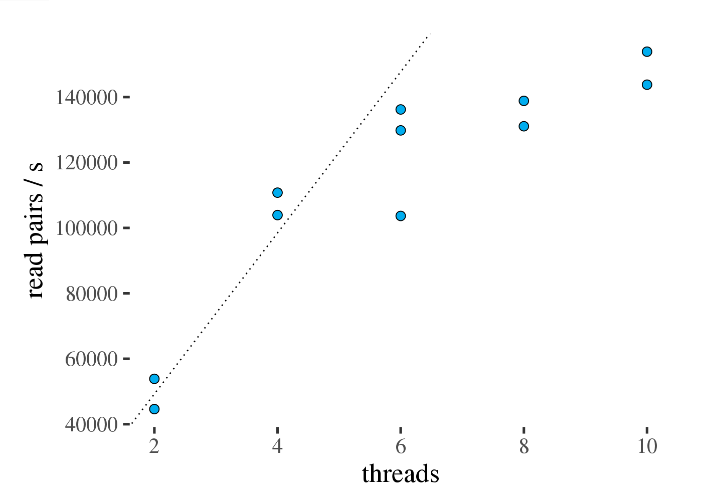
bwa mem 序列比对
比对过程实际上是拿着参考基因组,然后把每个小片段进行对齐,举个比较抽象的例子,就好比是拼图的过程,目的是让每一个小碎片都找到匹配的位置。
使用bwa mem程序进行比对,注意这里每个样品需要两条序列(因为二代双端测序两个方向是一对儿)。
bwa mem -M -t ${threads} \
-R "@RG\tID:$id\tPL:ILLUMINA\tLB:$lb\tSM:$sm" \
$genome \
/lscratch/$SLURM_JOBID/$(basename ${r1[small]}) \
/lscratch/$SLURM_JOBID/$(basename ${r2[small]}) \
> /dev/null
通过下图可以看出线程数与计算效率的关系,很明显在32线程以内基本呈正相关,因此建议此处设置的线程数稍微高一点,理论上线程数越多的话,拼图的速度也越快,就好比你找了几个小伙伴帮你同时拼图。
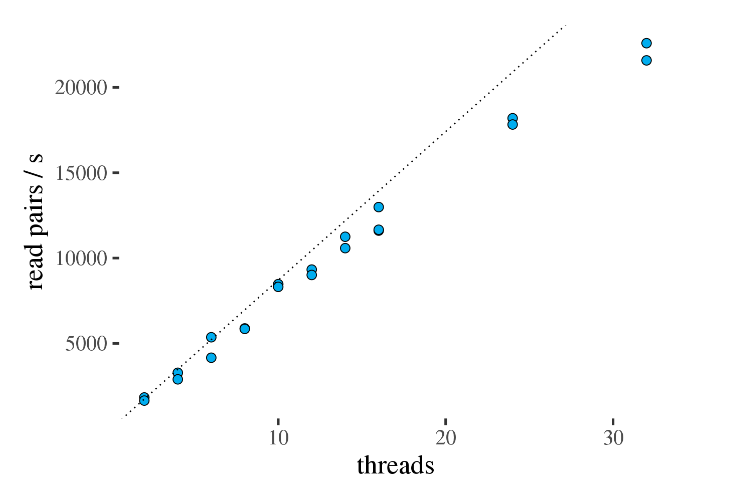 但是,也需要注意不要设置的太高,否侧多个线程的并行过程可能出现干扰,反而影响速度。就好比你为了拼一张地图把全校的人都叫过来了,一间屋里全是人,每个人移动起来都费劲,CPU也一样,线程过多相互打架。
但是,也需要注意不要设置的太高,否侧多个线程的并行过程可能出现干扰,反而影响速度。就好比你为了拼一张地图把全校的人都叫过来了,一间屋里全是人,每个人移动起来都费劲,CPU也一样,线程过多相互打架。
samtools sort 排序
SAM文件输出后直接管道传递给sort进行排序,这样可以避免磁盘读写浪费时间(就好比买了一份外卖我直接吃就行了,没必要拿到厨房加工一下再取出来,多此一举浪费时间)。可以再理解一下磁盘吞吐和溢出概念,这对提高性能非常重要。
使用重定向把输出信息丢弃,基准测试过程中BAM文件大概25GB左右,临时文件储存在lscratch中,需要格外注意设置线程数和每个线程的内存大小时综合考虑,需要根据机器配置和计算需求灵活修改。
samtools sort -T /lscratch/$SLURM_JOBID/part \
-m ${mem_mb_per_thread}M \
-@ ${threads} \
-O BAM \
/lscratch/$SLURM_JOBID/$(basename $bam) \
> /dev/null
通过对比可以发现,在设置10个线程时,计算效率最高,盲目的增加线程数反而导致计算效率降低了。另外,启用10个线程之后,设置内存参数也对速度有影响,例如16线程状态下,采用10GB内存的速度比30GB明显要快。 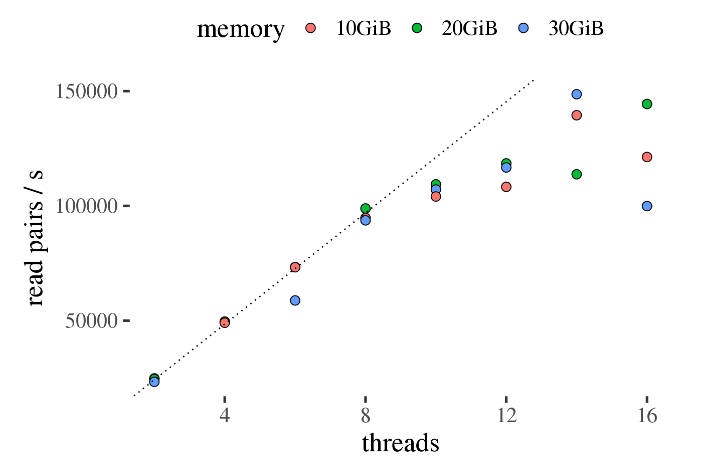 为了方便理解,举个不太抽象的例子:假如有很多乒乓球,每个上面有一个数字编号,打乱后放在一个屋子里,想让你按顺序排好放到另一个屋子。
为了方便理解,举个不太抽象的例子:假如有很多乒乓球,每个上面有一个数字编号,打乱后放在一个屋子里,想让你按顺序排好放到另一个屋子。
如果人数增加很明显是有助于提高排序速度(线程数增加),正常情况下每个人一次只能拿一个球(单个线程内存比较小),如果让每个人每次可以多拿几个球就可以避免来回跑(增加内存也能提高效率)。
但是存在某个临界情况,此时整体效率最大,也就是线程数和内存最匹配(上面图中的最高点处)。
samtools view转换bam文件
上步生成的sam文件要转换成bam文件,使用如下命令,此步骤占用的内存相对较少。
samtools view -@ ${threads} \
-O BAM \
/lscratch/\$SLURM_JOBID/$(basename $sam) \
> /dev/null
通过下图发现这一步设置的线程数和计算速度呈正比关系,测试最大采用16线程。 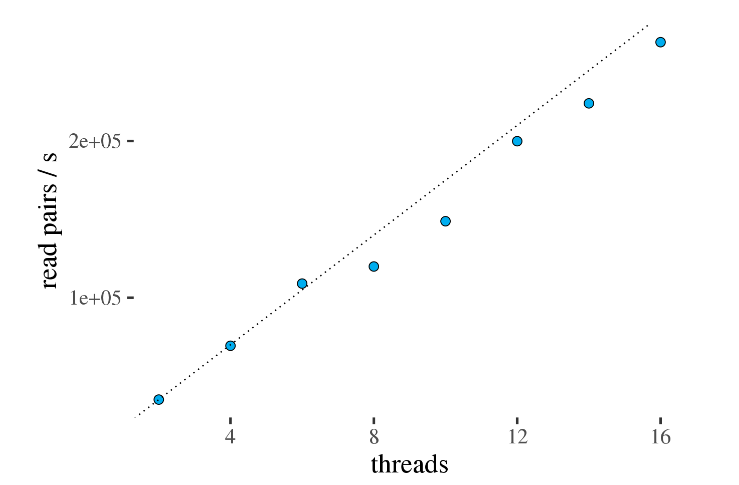
脚本的优化过程
有没有一种更厉害的方法,能达到比较高的计算效率呢?
下述代码中,使用两个管道命令将中间结果依次传递,采用32核心进行计算,完成了fastp、bwa、samtools等步骤。
fastp -i $read1 -I $read2 \
--stdout --thread 2 \
-j "${logs}/fastp-${SLURM_JOB_ID}.json" \
-h "${logs}/fastp-${SLURM_JOB_ID}.html" \
2> "${logs}/fastp-${SLURM_JOB_ID}.log" \
| bwa mem -v 2 -M -t 32 -p \
-R "@RG\tID:$id\tPL:ILLUMINA\tLB:$lb\tSM:$sm" \
$genome - 2> ${logs}/bwa-${SLURM_JOB_ID}.log \
| samtools sort -T /lscratch/$SLURM_JOBID/part \
-m 1706M \
-@ 12 \
-O BAM \
-o /lscratch/$SLURM_JOBID/$bam \
2> ${logs}/samtools-${SLURM_JOB_ID}.log
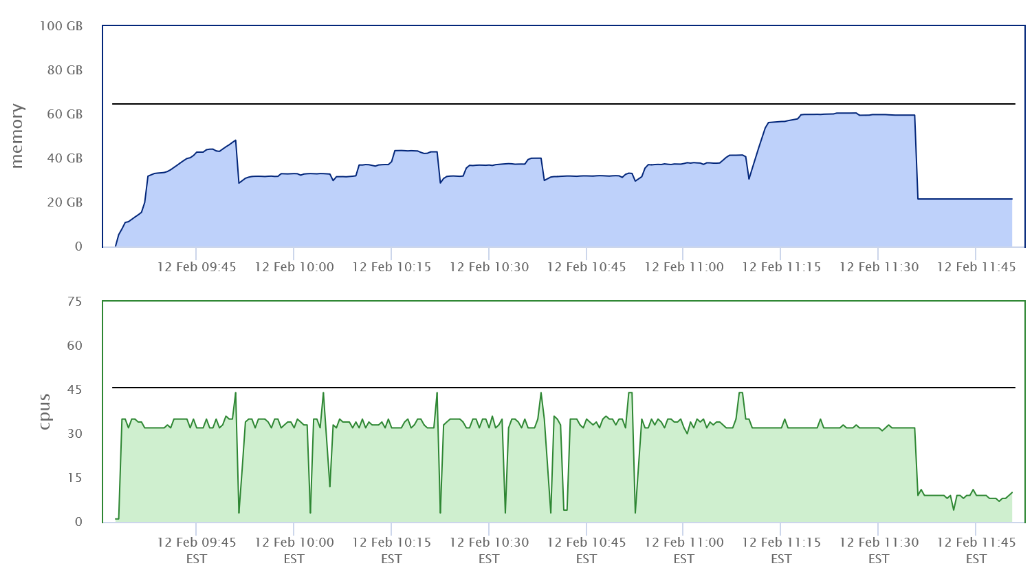
运行中占用的最大线程数为45左右,最大内存为60GB左右,大部分时间都是32线程进行运算,比对结果逐步记录到缓存区,当缓存区满了后再进行排序,这样极大地提高了效率。
本文由 mdnice 多平台发布















 5257
5257











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











