







摘要:RNA 结合蛋白 (RBP) 在活细胞的多个生物过程中发挥着重要作用,例如基因调控和 mRNA 定位。几种深度学习方法,特别是基于卷积神经网络(CNN)的模型,已被用于预测结合位点。然而,以前的方法无法代表RNA二级结构特征。传统的深度学习方法一般将RNA二级结构转化为规则矩阵,无法揭示RNA的拓扑结构信息。为了有效提取RNA的结构特征,我们提出了一种基于图卷积神经网络(GCN)和卷积神经网络(CNN)的RNA二级结构表示网络(RNASSR-Net),用于RBP结合预测。 RNASSR-Net构建源自RNA二级结构的图模型来学习RNA的拓扑特性。然后,通过CNN获得RNA中每个碱基的空间重要性,以指导RNA二级结构的表示。最后,RNASSRNet 结合结构和序列特征来预测结合位点。实验结果表明,所提出的方法在基准数据集上优于一些最先进的方法,并且在小规模数据上获得了更高的改进。此外,所提出的RNASSR-Net还用于检测与实验验证的基序相比的准确基序,这揭示了结合区域的位置和RNA结构解释,为未来的一些生物学指导。
1.介绍
RNA和蛋白质是生命的重要组成部分,参与活细胞的生化反应(Ule and Rinn 2014)。 RNA 结合蛋白是与 RNA 结合以调节基因表达并控制细胞内 RNA 过程和翻译的蛋白质(Van Nostrand et al. 2016)。基因调控包括生物体中大量的共转录和转录后基因表达,包括聚腺苷酸化、RNA剪接、修饰、加帽、定位、翻译和周转(Ray et al. 2013)。某些RNA结合蛋白(RBP)的功能障碍可能会导致一些严重的疾病,例如神经退行性疾病、癌症和心血管疾病(Musunuru 2003;Lukong et al. 2008。
RBP 对病毒 RNA 转录和复制也有很大影响。 2019 年冠状病毒病(COVID-19)的爆发是一种由新型冠状病毒引起的疾病。冠状病毒内部的冠状病毒 (CoV) N 蛋白在病毒 RNA 基因组包装过程中构建螺旋核糖核蛋白,调节复制/转录中的病毒 RNA 合成,并调节受感染的细胞代谢(Nelson、Stohlman 和 Tahara 2000;Stohlman 等人 1988)丛等人,2020)。因此,RBPs的研究有利于理解基因调控以及一些遗传性疾病和传染病的治疗。
为了研究 RBP 如何影响 RNA 加工,对每个 RBP 相互作用的 RNA 底物进行了分析(Ule et al. 2003)。这些RNA底物的结合位点与RBP的功能高度相关。 RNA 序列和 RNA 结构都决定了 RBP 结合强度(Buckanovich 和 Darnell 1997;Hackerm ̈ uller 等人 2005)。 RNA序列反映碱基分布信息,RNA结构反映拓扑信息。在之前的工作中,RNA序列被广泛用于获得RNA结合预测(Zhang et al. 2016;Kazan et al. 2010a)。然而,RNA结构的缺失并不能保证模型得到最优的预测。例如,含有“UGGC”的发夹环已被证明能够以高亲和力结合 Vts1p-SAM(Aviv 等人,2006)。因此,RNA结构提取的拓扑信息也有助于RNA结合预测。
为了分析RNA的结合,生物学家利用RBP通过生物学实验获得RNA的结合信息。然而,这种实验方式非常耗时、耗材。为了降低成本,高通量技术已广泛应用于RNA-蛋白质相互作用的基因组研究,例如交联免疫沉淀结合高通量测序(CLIP-seq)(Anders et al. 2011;Ferre) 、Colantoni 和 Helmer-Citterich 2015)和 RNAcompete(Ray 等人 2013)。这些高通量技术提供了大量可用数据,使得传统机器学习和深度学习方法能够训练准确的模型进行结合预测。
众所周知,RNA数据被视为序列,忽略了RNA的拓扑结构。结构数据可以通过一些计算软件直接获得,这有利于RBPs结合的研究(Gruber et al. 2008)。对于RNA二级结构,它表示为带有点和括号的简单字符串。这些点括号字符串使得卷积神经网络 (CNN) 和长短期记忆 (LSTM) 等模型很难学习绑定模式。一些方法根据RNA二级结构的核苷酸类别将点括号串转换为one-hot矩阵。然而,这种转变会导致RNA结构不可逆的信息丢失。因此,开发一种RNA二级结构分析方法迫在眉睫。
因此,我们提出了一种基于图卷积神经网络(GCN)和卷积神经网络(CNN)的RNA二级结构表示网络(RNASSR-Net),用于RBP结合预测。 RNASSR-Net 同时使用序列和结构数据来学习 RNA 的拓扑和结合特性,如图 1 所示。我们的源代码可在 https://github.com/ziniBRC/RNASSR-Net 上获取。本文的主要贡献包括以下内容
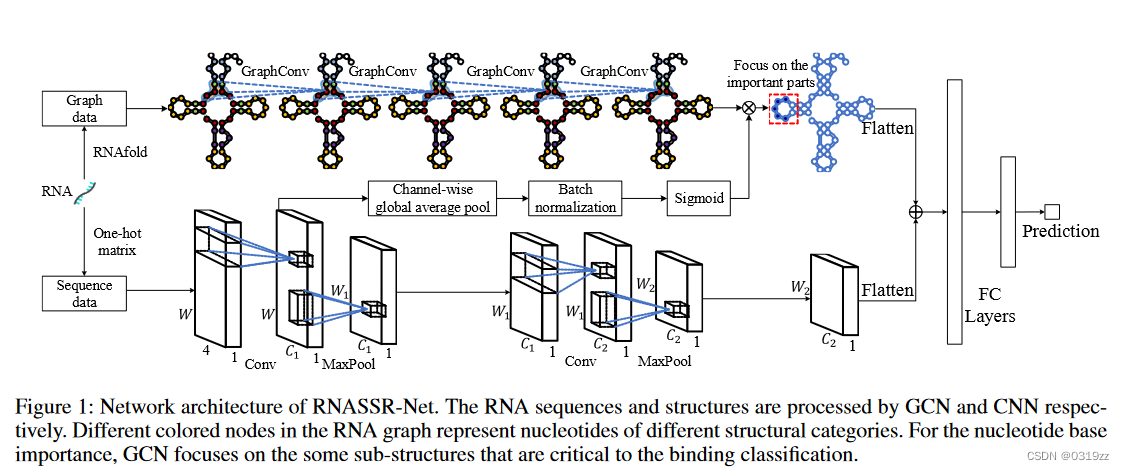
- 提出的RNASSR-Net模型使用GCN来表示RNA的二级结构,并使用CNN来学习RNA的序列特征。该方法同时考虑RNA的拓扑和序列特性,实现不同信息的互补,提高RNA结合的预测准确性。
- 我们使用 CNN 来学习揭示 RNA 空间重要性的碱基权重。权重可以指导 GCN 学习 RNA 的二级结构。碱基权重对应于 RNA 结构图中的节点。在相似的感受野下,我们共享碱基权重,使GCN专注于一些重要的核苷酸碱基。
- 我们融合结构和序列特征来预测 RBP 结合。所提出的方法优于基准数据集上的其他基线。我们检测到与实验验证的图案一致的准确图案。此外,我们分析每个核苷酸碱基的响应值以提取重要区域和结构突变信息。
2.相关工作
人们提出了许多传统的机器学习方法来解决 RNA 结合预测问题。传统方法关注如何手动提取重要特征。这些机器学习算法和计算工具分析特征的不同方面,以预测 RBP 结合位点并生成基序。 BioBayesNet 是第一个使用结构特征来执行转录因子结合位点目标识别问题的工具。 RNAContext 是一种基序结合方法,用于实现 RBP 对 RNA 序列和结构的相对结合偏好(Kazan 等人,2010b)。 RCK开发了一种新模型,该模型来自RNAcontext的扩展,以获得RNA结合蛋白的k-mer序列和结构偏好(Orenstein, Wang, and Berger 2016)。 GraphProt通过图编码从序列和结构信息中提取大量特征,并使用支持向量机(SVM)家族来预测RNA结合位点(Maticzka et al. 2014)。
由于RNA序列的复杂性,通过传统的机器学习方法很难提取重要的潜在特征。为了解决这个问题,深度学习被引入生物信息学和计算生物学领域,例如蛋白质定位(Almagro Armenteros et al. 2017)、蛋白质结构预测(Heffernan et al. 2015)、RNA预测(Alipanahi et al. 2015)。 2015)和化学信息学(Lusci、Pollastri 和 Baldi 2013)。最近,已经开发了几种深度学习方法来分析 RNA 结合预测并自动检测候选基序。 DeepBind(Alipanahi et al. 2015)首先引入CNN模型来预测RNA结合位点和结构。在 DeepBind 中,位置权重矩阵(PWM)由一层卷积和池化层以及全连接层组成,被嵌入到 CNN 模型中以显示结合位点的概率分布。 Deepnet-rbp(Zhang et al. 2016)使用深度信念网络(DBN)根据序列、二级和三级结构特征预测 RBP 结合位点。 iDeepE(Pan and Shen 2018)集成了多通道局部和全局序列信息,用于预测 RBP 结合位点和基序。另一项类似的工作 iDeepS 训练两个单独的 CNN 和一个 LSTM 来获得结合预测。虽然这些方法有效且准确,但它们主要关注使用 CNN 模型的 RNA 序列特征。根据RNA结构的类别将RNA结构的特征转换为位置权重矩阵,并由CNN模型处理。由于转换后RNA碱基的链接信息丢失,RNA结构的拓扑信息没有得到充分利用。
近年来,图神经网络(GNN)被发展来学习数据的拓扑信息,广泛应用于社会科学(Kipf and Welling 2017;Monti et al. 2019)、知识图谱(Schlichtkrull et al.)等领域. 2018;Chami 等人,2020),化学(Duvenaud 等人,2015;Gilmer 等人,2017)。为了处理不规则的数据格式,递归神经网络被用来构建图神经网络(Gori, Monfardini, and Scarselli 2005; Scarselli et al. 2008),其目的是处理一般类别的图。在固定大小图的情况下,一系列基于图的谱表示的卷积神经网络被应用于节点分类和图分类。具体来说,Kipf & Welling(Kipf and Welling 2017)提出了一种使用 1 跳滤波器的简化谱神经网络来解决过拟合问题并最小化操作数量。众所周知,很少有图神经网络被应用于RNA结构的分析。最近,基于 GNN 的 RNA-蛋白质相互作用网络 (RPI-Net) 用于学习和利用 RNA 分子的图形表示(Yan、Hamilton 和 Blanchette 2020)。
3.方法
3.1序列编码
RNA由四种核苷酸组成,可以通过核苷酸碱基的不同来区分。在我们的模型中,标记为“A”、“C”、“G”、“U”的碱基分别代表不同种类的核苷酸。数据集中RNA序列的长度不同。然而,卷积神经网络的输入特征必须具有相同的固定大小。我们使用标识符“N”将 RNA 序列填充到固定的窗口大小。给定一个 RNA 序列 s 作为 具有 n 个核苷酸 (
‘A’,‘C’,‘G’,‘U’,‘N’),我们将 RNA 序列数据编码为独热矩阵:

其中 i 是 RNA 序列中核苷酸的位置。在我们的研究中,我们假设 4 种不同的核苷酸在序列的开头和结尾服从平均分布。因此,我们对填充的核苷酸使用 [0.25, 0.25, 0.25, 0.25],在 one-hot 矩阵中使用“N”。 “N”表示 RNA 序列中的未知碱基。
3.2图构建
one-hot 矩阵反映了核苷酸序列的组成。然而,RNA 的结构在 RNA 功能中起着重要作用。 RNA序列的One-hot矩阵不能反映核苷酸结构信息。为了更好地提取RNA的内在特性,模型中应考虑RNA结构。在本文中,我们使用 RNAfold 来抽象某些结构细节。 RNAfold 可以对所有可能的结构进行采样并保留高度可能的候选结构。在以前的几种方法中,模型中使用了二级结构。二级结构中的核苷酸可分为六类,分别表示为茎(S)、多环(M)、发夹(H)、内环(I)、凸起(B)和外部区域(E)。与RNA序列相同,这六类核苷酸可以在之前的方法中转移到one-hot矩阵中。然而,one-hot矩阵忽略了RNA的结构关系,可能会丢失一些结构信息。因此,我们使用图模型来表示RNA的结构信息。
根据RNAfold提取的结果,一些结构可能在两个核苷酸i和j之间具有连接,但其他结构可能没有。在这种方法中,我们将与序列 x 相关的二级结构 s 的概率表示为 。 (Yan, Hamilton, and Blanchette 2020) 中也使用了图的构造。概率
定义为:
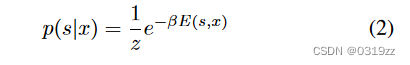
其中 Z 是归一化常数,E(s, x) 是结构 s 下 x 的自由能。核苷酸 i 和 j 的碱基配对概率定义为:

运行 RNAplfold 后,我们获得概率邻接矩阵 ,其中
。对于不同RNA序列构建的图,图的节点数也不同。我们使用未链接到其他节点的空节点填充图表。填充图的节点数与填充RNA序列的长度相同。
3.3网络架构
对于RNA数据,结构和序列可以表示为图数据和one-hot矩阵。由于之前基于 CNN 的方法不用于图数据,因此我们提出了一种基于 GNN 和 CNN 的新型网络来学习 RNA 的结构和序列特征。该网络整合了图中的节点信息和one-hot矩阵中的空间信息来进行RNA结合预测。在网络中,图数据和one-hot矩阵分别输入到GNN和CNN中。 GNN 和 CNN 都有两个卷积层和两个池化层。对于图数据和one-hot矩阵,一个核苷酸对应于图中的一个节点和one-hot矩阵中的一个位置。因此,RNA图中的节点和RNA序列中的核苷酸碱基应该共享一些相似的信息。在相似感受野下,将CNN提取的空间重要性传递给GCN,指导GCN的训练过程。最后,将GCN和CNN提取的特征进行展平,将其相互串联。然后,将这些特征输入到全连接层中以获得 RNA 结合预测。
3.3.1卷积神经网络
卷积神经网络(CNN)可以帮助提取输入的非线性内在特征,包括卷积、最大池和全连接层。任何卷积神经网络都至少包含一个卷积运算。卷积运算后输出输入的one-hot矩阵和滤波器之间的逐点乘积。卷积运算 的输出是与填充序列 s 中的核苷酸 i 对齐的滤波器 k 的分数。在本研究中,滤波器存储在矩阵 F 中,其中元素
是第
层中位置 j 和通道 c 处的第 k 个滤波器的系数。卷积运算的输出
可以定义为:
![]()
其中 是第
层的输出,i 是特征索引,
是第
层中滤波器 k 的系数,m 是滤波器的大小,
是第
层输入的通道数。在第一层的卷积运算中,卷积运算的输入是独热矩阵M,M的通道是核苷酸的基数。因此第一个卷积滤波器被称为基序检测器,它可以学习碱基分布的模式。
卷积运算的激活函数是修正线性单元(ReLU)。大于0的正分数被传递到下一层,而负分数被分配为0。ReLU激活操作定义为:
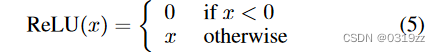
在 CNN 中,卷积运算后面是最大池层或平均池层。经过卷积层之后,输出特征图的大小几乎保持不变。池层可以通过选择特定窗口内的最大值或平均值来降低维度并保留一些重要特征。最大池层可以表示为:
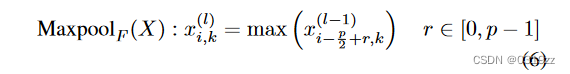
其中 X 是卷积和 ReLU 层之后的输出,p 是池化层核的长度,是
最大池化层的输出,该输出将输入到下一个卷积层或全连接层。
3.3.2图卷积神经网络
图神经网络(GNN)是一种学习分层图表示和图节点嵌入的端到端网络。在本文中,我们使用GNN学习相邻节点信息和节点嵌入特征来分析RNA结构。对于之前的方法,构建RNA结构的one-hot矩阵并将其输入到卷积神经网络中进行预测。然而,将RNA结构转化为one-hot矩阵的过程会丢失一些重要的信息。此外,one-hot矩阵只能分析节点特征,往往会忽略相邻节点关系。而GNN可以直接处理结构数据。将相邻矩阵输入到GNN模型中,提取相邻节点信息。
给定图 G = (V, E,A),其中 V 是 |V| = n 节点的有限集,E 是边集, 是编码两个节点之间的连接权重的邻接矩阵。为了便于理解,我们考虑遵循一般“消息传递”架构的图卷积:
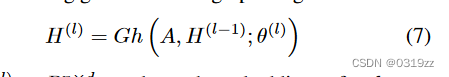
其中是经过
步图卷积运算后的节点嵌入,Gh(·) 是图卷积运算,称为消息传播函数,
是最后一个卷积运算的输出,
是可训练参数。初始
是RNA二级结构的节点特征。
为了获得图卷积运算的输出,已经提出了许多消息传播函数的实现。一种流行的方法是图卷积网络,它是通过线性变换和 ReLU 非线性实现的:
![]()
其中 且
是可训练矩阵。
是
的度矩阵。
是一种重整化技巧,旨在缓解数值不稳定和梯度爆炸/消失问题。
尽管 GCN 已应用于许多研究领域,但 GCN 并没有产生很好的预测性能。在本文中,我们介绍了一种新方法,可以利用 CNN 的特征来帮助 GCN 表现更好。在RNA结构的图学习表示中,RNA序列中的每个碱基对应于图中的一个节点。在相似的感受野下,CNN学习到的基重要性权重可以指导GCN的训练过程。对于一个图卷积层来说,节点整合了邻居节点的信息,可以理解为CNN中的3*1滤波器。对于常规数据,4 个图卷积层与 CNN 中的 9*1 滤波器相比具有相似的感受野。因此,我们从第一个 CNN 层提取基本重要性权重来指导 GCN 的训练过程。基本重要性权重定义为:
![]()
其中是第一个CNN层的输出,BN是批量归一化的操作。这个重要性权重乘以GCN得到的图特征,计算如下:
![]()
其中 H(4) 是第 4 个 GCN 层的输出,是 Hadamard 乘积。
3.3.3损失函数
模型的输出用于预测 RNA 数据的结合标签。为了获得最优参数,我们使用二元交叉熵损失函数:
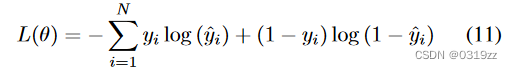
其中是真实标签,
是训练模型的预测标签。
3.4基序检测
在我们的研究中,我们将采用 MEME(Bailey et al. 2009)来完成我们结果的可视化。在本研究中,我们研究了模型中 CNN 和 GCN 的卷积输出。卷积滤波器可以被视为“图案检测器”。如果过滤器具有相同的基序碱基分布,则这些过滤器的输出反映结合强度。我们使用 DeepBind 和 Basset 中相同的策略将滤波器的输出转换为位置权重矩阵(PWM)(Alipanahi 等人,2015)。我们计算 CNN 中第一个卷积的输出和 GCN 中最后一个图卷积的输出的平均值。如果第 i 个位置的平均输出值大于这组序列的最大值 0.75,则会选择 k-mer 序列来计算图案相应位置上每个碱基的概率。
4.实验
4.1基线方法
已经开发出许多计算方法来从序列数据集中预测结合位点和基序结构。在本研究中,我们将我们的方法与其他四种最先进的方法进行比较,并通过接受者操作特征曲线下的面积(AUROC)来衡量性能。
- GraphProt(Maticzka et al. 2014):它使用了一个图内核,可以表示序列特征和潜在主题的潜在二级结构。这些图内核特征被输入到支持向量机(SVM)中,以对结合位点和未结合位点进行分类。
- Deepnet-rbp(Zhang et al. 2016):它通过将RNA一级序列、二级和三级结构特征融合在一起来构建特征,并将该特征输入到DBN中以预测RBP结合位点。
- iDeepE(Pan and Shen 2018):一方面,它使用两层局部多通道CNN对RNA序列的多个子序列进行并行卷积以获得局部输出。另一方面,它使用两层全局 1 通道填充 RNA 序列来获得全局输出。最后,取局部和全局输出的平均值作为最终输出。
- RPI-Net(Yan、Hamilton 和 Blanchette 2020):提出了一种基于门控 GNN 的模型来学习 RNA 二级结构的图形表示。它使用 LSTM 将节点嵌入视为隐藏状态,并将到达每个节点的消息视为输入。
4.2数据集
我们将我们的方法的性能与最初由(Maticzka 等人,2014)创建的 GraphProt 中的基准 RBP 绑定数据集的其他基线进行比较。该数据集由 24 组 HITS-CLIP、PAR-CLIP 和 iCLIP 衍生的结合位点组成,其中 23 组来自 doRiNA (Anders et al. 2011),PTB HITS-CLIP 结合位点取自 (Xue et al. 2011)。 2009)。对于该数据集,CLIP-seq 实验中识别的区域两端各有 150 个核苷酸。根据阳性位点,通过实验鉴定出每个RNA阳性序列中的“视点”区域。阳性位点是锚定在峰中心的子序列,这些子序列源自在 doRiNA 中处理的 CLIP-seq。阴性 RNA 样本是通过随机选择人类转录本视点大小的部分来生成的,其中没有现有结合位点的支持证据。
根据 CLIP-Seq 协议,包括 PARCLIP (Hafner et al. 2010) 和 HITS-CLIP (Licatalosi et al. 2008),使用特定的 RNA 切割酶(例如核糖核酸酶 T1。由于相同的裂解酶,RNA区域的边界具有相同的碱基分布,这可能会误导模型学习裂解酶的信息。为了避免边界误导问题,通过使用四种核苷酸随机化 RNA 区域的边界来对数据集进行去偏。
在本文中,我们仍然关注原始绑定数据集。然而,RPI-Net 不提供原始数据集的结果。因此,我们对去偏数据集进行了一些扩展实验,以将 RNASSR-Net 与 RPI-Net 进行比较。
4.3参数设置
为了研究我们模型的性能,我们随机选择 RBP-24 中 90% 的原始训练集作为训练集,剩下的 10% 作为验证集。我们使用 Adam 对模型进行最多 100 个时期的训练(Kingma 和 Ba 2015)。对于每个epoch,我们将批量训练数据的大小设置为128。对于学习率参数,我们将初始学习率设置为0.001,学习率降低因子设置为0.5。如果验证损失连续 10 个 epoch 没有减少,则学习率将按学习率减少因子减少。当学习率等于1e-5时,训练过程将停止。当模型在验证数据上获得最高 AUC 时,我们保存模型。其他参数的微小变化并没有对结果产生太大影响。我们将权重衰减和 dropout 分别设置为 0.01 和 0.25。对于基线模型,我们设置与原始论文相同的参数。
我们在配备 NVIDIA GTX 2080Ti GPU、内存 12 GB 的 Ubuntu 服务器上运行我们的实验。初始权重和偏差使用 PyTorch 中的默认设置。我们使用 DGL 构建图,DGL 被称为图神经网络的开源框架。
4.4将预测结果绑定到原始数据集上
我们将我们的方法与 24 个蛋白质子数据集上的其他基线方法进行比较。详细结果列于表1中。总的来说,我们的方法在几乎所有蛋白质上都比其他基线方法表现更好,这证明我们的方法可以在预测RNA结合位点的任务中表现更好。
如表 1 所示,我们的方法在 24 个子数据集上产生了最佳平均 AUC 94.4%,比 GraphProt 好 5.7%,deepnet-rbp 好 4.2%,ideepE 好 1.3%。 GraphProt 和 Deepet-rbp 是序列结构轮廓方法,而 iDeepE 和我们的方法是 PWM 轮廓模型。 iDeepE 和我们的方法可以更好地自动从神经网络中的卷积核中学习潜在的主题。 iDeepE 仅从序列数据中提取 RNA 特征,而我们的方法使用 GCN 集成 RNA 结构特征。 RNA结构的引入大大提高了结合预测。此外,GraphProt 的性能比其他方法更差。支持向量机(SVM)是一种传统的机器学习方法,在 GraphProt 中用于预测结合位点的出现。尽管GraphProt使用图内核来表示RNA结构,但图内核无法充分表示用于预测的结构信息。从结果来看,深度学习方法相对于传统机器学习方法可以表现出明显的优势。 Deepnet-rbp利用DBNs提取RNA二级结构和三级结构特征,但维度越高,RNA结构的准确性越低。 Deepnet-rbp对RNA结构数据进行编码,这意味着信息缺失。我们的方法选择表示为图数据的原始RNA结构进行预测,以尽可能多地提取有价值的信息。在 4 种方法中,我们的方法对 21 种蛋白质产生了最佳 AUC。对于另外三个蛋白质数据集,deepnet-rbp 获得了比其他方法最好的 AUC,而我们的方法具有与 deepnet-rbp 非常相似的 AUC。
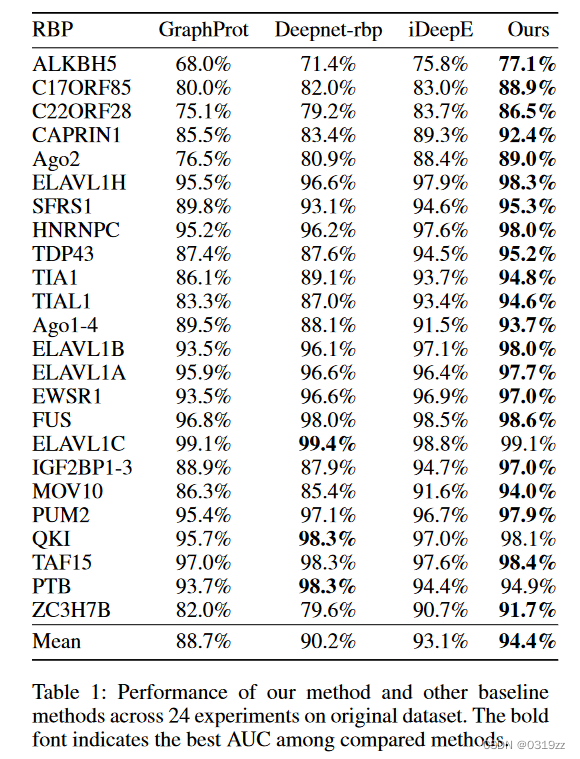
此外,我们的方法显着提高了小规模子数据集的性能。与最佳基线 AUC 相比,我们的方法使 ALKBH5 增加 1.3%,C17ORF85 增加 5.9%,C22ORF28 增加 2.8%,CAPRIN1 增加 3.4%。
4.5在去偏数据集上结合预测结果
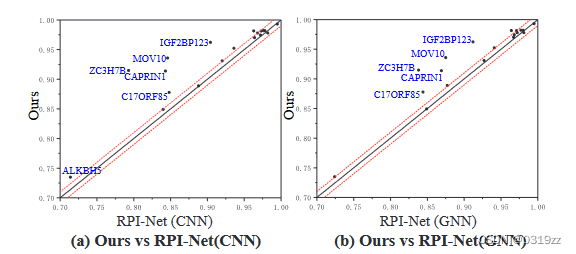
对于去偏数据集,正样本中“视点”区域的边界是随机的。从之前的研究来看,基于CNN的方法的性能会因为酶切信息的丢失而下降。为了确保我们的方法能够在没有酶裂解信息的情况下学习预测模式,我们对 19 个去偏子数据集进行了一些比较实验。具体来说,我们的方法在去偏数据集上优于其他基线方法。我们的方法在 19 个去偏子数据集上产生了最佳平均 AUC 93.6%,比 GraphProt 好 7.3%,deepnetrbp 好 3.9%,ideepE 好 5.8%,RPI-Net 好 1.9%。
RPI-Net 在去偏数据集上比其他基线方法获得更好的性能。因此,我们在图2中展示了我们的方法和RPI-Net之间的比较。我们的方法总体上优于RPI-Net,因为二级结构和序列的整合特征可以提高RNA结合预测的性能。与 RPI-Net 相比,我们在几个 RBP(ALKBH5、C17ORF85、CAPRIN1、SFRS1、ELAVL1 和 ZC3H7B)上的方法实现了超过 1% 的 AUC,这表明二级结构特征的重要性。RPI-Net 在去偏数据集上比其他基线方法获得更好的性能。因此,我们在图2中展示了我们的方法和RPI-Net之间的比较。我们的方法总体上优于RPI-Net,因为二级结构和序列的整合特征可以提高RNA结合预测的性能。与 RPI-Net 相比,我们在几个 RBP(ALKBH5、C17ORF85、CAPRIN1、SFRS1、ELAVL1 和 ZC3H7B)上的方法实现了超过 1% 的 AUC,这表明二级结构特征的重要性。
4.6可视化
与 RBP 的其他深度学习方法一样,我们的方法可以从模型中第一个卷积滤波器的学习参数中自动识别结合序列图案。经过多次实验,我们在 RBP-24 数据集中得到了 RBP 的候选主题。我们对一些已识别的motif进行采样,以显示图3中RNASSR-Net的motif检测能力。根据当前CISBPRNA和其他一些研究的知识,我们计算了序列中候选motif的频率,并显示了已知的motif和检测到的motif。我们使用 TOMTOM 的方法的主题。
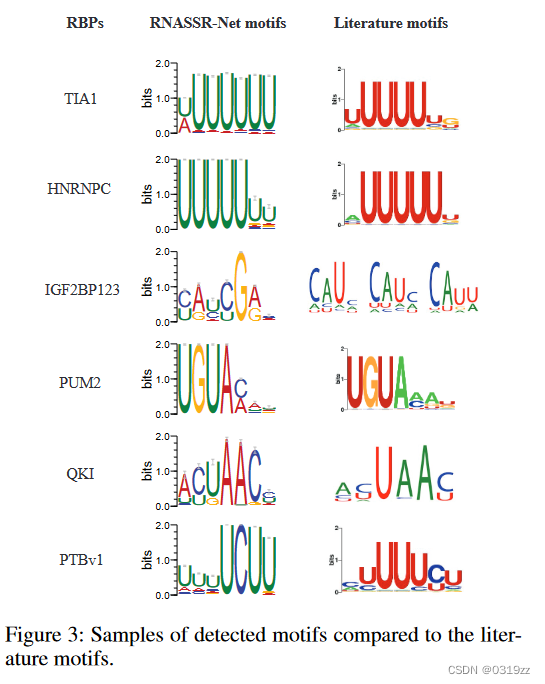
然而,有许多蛋白质在当前的 CISBP-RNA 中没有相应的经过验证的基序。因此,我们进行富集分析以显示这些 RBP 最可能的基序。我们将这些检测到的主题与文献中的一些数据进行比较。 FUS、TAF15 和 EWSR1 显示出对富含 AU 的位点的偏好(Hoell 等人,2011)。
4.7RNA 响应分析
我们新颖地从图4中的CNN和GCN的特征中提取RNA中每个核苷酸的响应值。峰值区域是序列中预测的“视点”,这有助于结合预测。曲线下的序列对应于碱基的序列类别和结构类别。特别地,GCN结果中具有最小自由能的结构是由RNAfold预测的。从图4中可以看出,峰面积与真实的“视点”区域一致,这表明RNASSR-Net具有良好的识别基序区域的能力。
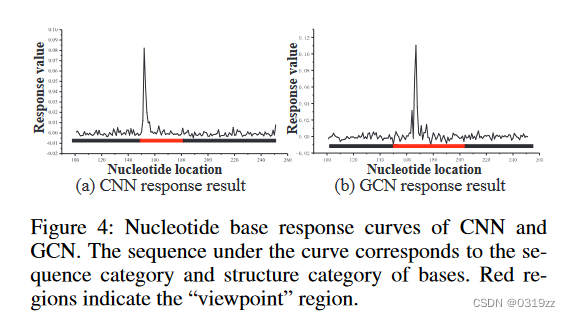
GCN的核苷酸碱基响应曲线可以反映结构突变。在图4(b)中,“视点”区域的左边界是“S”到“H”的过渡,其中存在曲线的波谷。在峰区,第一峰和第二峰之间存在一个谷,对应结构中的“SSBBSSS”。 “B”的突变导致两个峰之间的波动。
5.结论
在本研究中,我们开发了一种基于 CNN 和 GCN 的新型深度学习模型,用于根据 CLIP-seq 得出的数据预测 RBP 结合位点。为了指导 GCN 训练过程,我们将 CNN 提取的基本权重引入到 RNA 图的节点中。我们对原始数据集和去偏数据集进行了多次实验。从实验结果来看,我们的方法比其他最先进的方法具有更好的性能。 GCN 和 CNN 的集成改进了原始数据集和去偏数据集上的深度学习模型。此外,我们的方法可以自动检测准确的结合基序。我们新颖地分析了 CNN 和 GCN 的响应曲线,以解释 RNA 的聚焦区域和结构突变,以进行结合预测。















 694
694

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









