






|作者:张林峰1,2,† 王涵3,4,††
(1 北京科学智能研究院)
(2 北京深势科技有限公司)
(3 北京应用物理与计算数学研究所 计算物理全国重点实验室)
(4 北京大学工学院 应用物理与技术研究中心)
本文选自《物理》2024年第7期

摘要 随着人工智能技术的飞速发展,其与物理建模的结合为微观尺度的科学研究带来了革命性的工具。文章介绍了从薛定谔方程出发的量子力学近似求解方法到大原子模型(LAM)的发展历程,并特别关注机器学习技术在原子尺度模拟中的应用。文中首先讨论人工智能与物理建模结合的理论基础,随后深入分析这一结合在原子尺度模拟中的实现方式,包括机器学习模型的构建和训练策略。还探讨了数据积累、软件工具和工程基础设施对推动该领域进步的重要性,并展望了大原子模型在未来科学研究和工业应用中的潜在影响。通过不断的技术创新和跨学科合作,大原子模型将在材料科学、化学工程、生物技术等多个领域发挥重要作用,推动科学研究和工业应用进入新的发展阶段。
关键词 人工智能,物理建模,量子力学,大原子模型,原子尺度模拟

01
引 言
原子,这一构成物质世界的基本单位,虽眼不可见、手不可触,但与我们日常生活的各个方面都息息相关。从化学反应合成的药物用于治疗疾病,到通过材料科学改进的合金应用于航空航天,再到日常用品如清洁剂和塑料的制造,都基于对原子性质和相互作用的理解。美国物理学家理查德·费恩曼曾指出:“如果说有一个最强有力的假设能引导我们不断探索生命的奥秘,那便是所有事物均由原子构成,生物体的一切行为都可以用原子的振动与摆动来解释。”[1]从古希腊哲学家德谟克利特首次提出物质不可再分的最小单元——原子的概念,到19世纪门捷列夫构建元素周期表,人类对原子世界的探索已历经数千年。
从19世纪初到20世纪初,人类基本完成对原子世界的理论建模。1808年,约翰·道尔顿提出现代版本的原子理论,认为每种元素由一种独特的原子组成;1897年,约瑟夫·汤姆孙发现电子,并在1904年提出“葡萄干布丁模型”描述原子结构;1911年,欧内斯特·卢瑟福通过金箔实验提出原子中存在密集正电核的模型;1913年,尼尔斯·玻尔基于量子化概念提出电子沿特定轨道运动的模型,解释了氢原子光谱线;1926年薛定谔方程的提出标志着原子世界的理论建模基本完成(图1);1928年,结合量子力学与相对论的狄拉克方程,则改进了对重原子和光谱精细结构等的描述。1929年,狄拉克对量子力学的应用与挑战进行了深入的阐述:“……对大部分物理学和整个化学进行数学建模的基本物理定律已经完全知晓,而困难仅在于基于这些定律的数学方程过于复杂以致无法求解。因此,有必要发展量子力学的近似实用方法,这些方法在不需要太多计算的情况下,可以解释复杂原子系统的主要特征。”[2]
图1 从左至右:薛定谔(Erwin Schrödinger),狄拉克(Paul A. M. Dirac),玻恩(Max Born)
自薛定谔方程诞生以来的近百年间,“发展量子力学的近似实用方法”成为人们研究电子与原子、分子与材料世界的主旋律。在这个过程的大致前40年,科学家们主要依靠手工理论推导和简单的计算工具。之后50年,随着计算机技术的普及与发展,计算逐渐成为除理论和实验外的第三种主要科研手段,一系列理论和数值算法得到了迅速完善,使精确模拟每个原子的运动成为可能。最近10年,机器学习的引入以及大模型的出现,极大地扩展了人类模拟原子世界的时空尺度边界,相关方法已经成为科学家探索和应用原子世界规律的重要工具。
在本文中,我们以“发展量子力学的近似实用方法”为主线,着重介绍近10年出现的机器学习原子尺度模拟方法的现状与未来。为了讨论更为深入,我们首先简要介绍微观模拟的基本原理与数值方法,然后介绍建模过程中综合机器学习与物理的考量,紧接着介绍数据积累和打通应用的重要性,并探讨软件与工程基础设施的发展对这一领域的影响。文末,我们介绍最近两年来人们对原子尺度大模型的思考与探索,展望大原子模型在原子级设计、表征和生产制造方面的潜在应用,讨论它彻底改变材料科学、化学工程和生物技术等领域可能途径。

02
原子尺度模拟的量子力学模型
在不考虑相对论效应的前提下,对微观电子—原子核体系的建模始于量子力学的基础方程——薛定谔方程[3]。其不含时形式为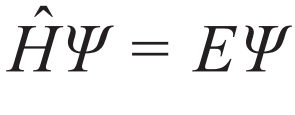 ,其中Ψ为波函数,
,其中Ψ为波函数, 为哈密顿量,E为体系能量。薛定谔方程的提出并不意味着基于第一性原理的建模方案的终结,而是一个开始。关键的挑战在于将量子力学原理应用于现实化学或更一般的工程问题时的数学复杂性。Walter Kohn在他著名的诺贝尔奖演讲中指出[4],当应用于多粒子系统时,传统的多粒子波函数方法会遇到所谓的指数灾难。在忽略自旋指标时波函数为体系中原子核和电子坐标的函数,例如在一个水分子的体系中,我们有2个氢原子核,1个氧原子核以及10个电子,则波函数为3×13=39维空间中的函数。即使每个维度只需要3个网格点进行离散,总网格数也将是339≈4×1018。仅仅以单精度存储这些数据就需要约18 EB (1 EB=10243 GB)内存。此外,还涉及电子—电子和电子—离子相互作用产生的高度纠缠,以及波函数必须满足在两个相同的电子交换位置时改变其符号的大名鼎鼎的泡利不相容原理[5]等等。为这个问题开发高效算法是计算科学中最英勇的尝试,并且取得了一定的成功。目前为止,开发的主要方法包括Hartree—Fock方法[6,7]、组态相互作用方法[8]、耦合簇方法[9]、量子蒙特卡罗方法[10],以及最近的密度矩阵重正化群理论[11]和密度矩阵嵌入理论[12]等。
为哈密顿量,E为体系能量。薛定谔方程的提出并不意味着基于第一性原理的建模方案的终结,而是一个开始。关键的挑战在于将量子力学原理应用于现实化学或更一般的工程问题时的数学复杂性。Walter Kohn在他著名的诺贝尔奖演讲中指出[4],当应用于多粒子系统时,传统的多粒子波函数方法会遇到所谓的指数灾难。在忽略自旋指标时波函数为体系中原子核和电子坐标的函数,例如在一个水分子的体系中,我们有2个氢原子核,1个氧原子核以及10个电子,则波函数为3×13=39维空间中的函数。即使每个维度只需要3个网格点进行离散,总网格数也将是339≈4×1018。仅仅以单精度存储这些数据就需要约18 EB (1 EB=10243 GB)内存。此外,还涉及电子—电子和电子—离子相互作用产生的高度纠缠,以及波函数必须满足在两个相同的电子交换位置时改变其符号的大名鼎鼎的泡利不相容原理[5]等等。为这个问题开发高效算法是计算科学中最英勇的尝试,并且取得了一定的成功。目前为止,开发的主要方法包括Hartree—Fock方法[6,7]、组态相互作用方法[8]、耦合簇方法[9]、量子蒙特卡罗方法[10],以及最近的密度矩阵重正化群理论[11]和密度矩阵嵌入理论[12]等。
为进一步高效求解更大尺度体系上关心的问题,人们探索了一系列从薛定谔方程出发的多尺度建模的方法。多尺度建模的核心目标在于通过结合不同层级的理论和计算方法,实现从微观到宏观各个层次体系的精确模拟。对此与机器学习结合的介绍可参考文献[13],本文只聚焦于原子尺度。首先,玻恩和奥本海默观察到[14],电子和原子核之间有明显的时间尺度分离:电子的运动总是远快于原子核,因此通常可以假设电子总是处于其基态,并随原子核进行绝热演化。这样,只需经典的牛顿运动方程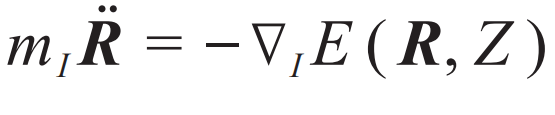 来描述原子的运动,其中E(R, Z)为势能,是所有原子核坐标R和类型Z的函数。注意R是所有原子核坐标的集合,对于一个水分子,共有三个原子核坐标,因此势函数在坐标自由度上是一个3×3=9维的函数。这里,我们暂时避免加入更多细节,如周期性边界条件或热浴,并把通过求解牛顿运动方程模拟原子运动的方法为分子动力学方法。
来描述原子的运动,其中E(R, Z)为势能,是所有原子核坐标R和类型Z的函数。注意R是所有原子核坐标的集合,对于一个水分子,共有三个原子核坐标,因此势函数在坐标自由度上是一个3×3=9维的函数。这里,我们暂时避免加入更多细节,如周期性边界条件或热浴,并把通过求解牛顿运动方程模拟原子运动的方法为分子动力学方法。
势函数E(R, Z)可以通过求解电子的不含时薛定谔方程获得。尽管量子化学领域取得了显著进展,但由于计算复杂度、计算精度、能否解析求导等原因,仍然很难将上述方法用于在分子动力学模拟中实时计算势函数及其梯度。在这种情况下,Kohn—Sham(KS)密度泛函理论(DFT)[15,16],由于其较好地平衡了效率和准确性,以及可以方便地计算梯度,在过去几十年中成为占据主导地位的薛定谔方程近似求解方法。在KS密度泛函理论框架下,假设电子处于基态,静态外部势中相互作用电子的棘手多体问题被简化为在有效势中的非相互作用电子的可处理问题,即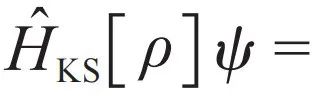
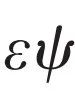 。薛定谔方程中多电子波函数Ψ被简化为单电子波函数ψ,这意味着求解问题的维度从三倍电子个数的维度降低到3维,从而成功地解决了维数灾难。值得注意的是,KS密度泛函理论的哈密顿算符成为了一个依赖于电子密度的算符,而电子密度直接由问题的解,单电子波函数ψ决定,因此问题从一个线性特征值问题转化为一个非线性特征值问题。KS密度泛函理论求解的计算复杂度由特征值问题求解决定,一般而言为电子数的三次方。KS密度泛函理论的哈密顿算符为
。薛定谔方程中多电子波函数Ψ被简化为单电子波函数ψ,这意味着求解问题的维度从三倍电子个数的维度降低到3维,从而成功地解决了维数灾难。值得注意的是,KS密度泛函理论的哈密顿算符成为了一个依赖于电子密度的算符,而电子密度直接由问题的解,单电子波函数ψ决定,因此问题从一个线性特征值问题转化为一个非线性特征值问题。KS密度泛函理论求解的计算复杂度由特征值问题求解决定,一般而言为电子数的三次方。KS密度泛函理论的哈密顿算符为
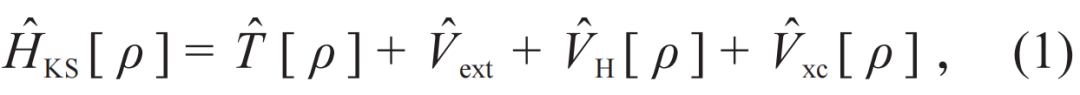
等式右边各项依次为动能算符、原子核—电子作用的外势算符、电子间库仑作用算符,这三个算符均有解析表达形式。最后一项为交换关联算符,它是人类为使用KS密度泛函理论付出的代价:我们只知道它的存在性而不知其解析表达形式。这意味着,虽然理论上KS密度泛函理论在电子基态问题中等价于薛定谔方程,但是实际求解精度完全依赖于交换关联算符的构造精度[17]。
基于KS密度泛函理论的分子动力学通常被称为从头算分子动力学(ab initio molecular dynamics,AIMD)[18]。尽管相比基于波函数的量子化学方法,AIMD的计算效率有了巨大提高,但其计算成本仍然使其受限于数百个原子和时间尺度为约10 ps (10-11 s)的典型应用,并且其内蕴的计算复杂度标度问题也限制了高性能计算系统(HPC)的有用性。例如从2006年到2019年,尽管世界上最快的超级计算机的峰值性能增加了550倍 (从BlueGene/L的360 TFLOPS(每秒1012次浮点运算)增加到Summit的200 PFLOPS (每秒1015次浮点运算)),但AIMD可以计算的最大系统仅增加了8倍,这几乎完美地遵循了立方标度的规律[19]。

03
机器学习势函数
随着近年来机器学习,特别是深度学习的迅猛发展,原子世界的研究迎来了新的机遇。在数据驱动的研究领域中,2020年AlphaFold-2的问世标志着一个重要的里程碑[20]。AlphaFold-2利用深度学习模型可以精准预测蛋白质的原子级三维结构,对结构生物学和药物设计等领域产生了革命性的影响。在由量子力学基本原理驱动的研究中,机器学习势函数的开发为分子动力学研究提供了新的可能[21]。2017年提出的DeePMD(深度势能分子动力学)方法通过利用深度学习技术构建势函数,在保持量子力学精度的同时将计算复杂度降低到线性标度[22]。2020年,DeePMD的开发团队结合了机器学习、物理模型和高性能计算,将人类进行量子力学精度的原子模拟能力从万原子提升到亿原子,且每日进行物理时间1 ns(10-9 s,注意模拟时间步长仅有10-15 s)的模拟,并因此获得高性能计算应用领域的最高荣誉——戈登贝尔奖[19]。这个阶段,深度学习带给研究者最为关键的能力,便是克服维数灾难、表示与逼近高维函数、处理大规模数据的能力。
3.1 模型构造与物理约束
势函数E(R, Z)是一个从所有原子核坐标R及其类型Z到势能E的映射,这个函数的坐标空间维数为三倍原子数。因此对于任何非平凡体系,势函数都是一个高维函数。Behler—Parrinello(BP)[21]在其开创性的工作中将势能分解为所有单原子能量(Eatom)贡献的求和形式,每个原子的能量贡献是原子局域环境的函数:
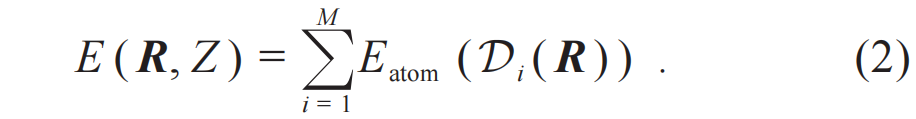
这里每个原子的局部环境是指,在三维空间中以某个原子为中心的球体内,所有“邻居”原子的元素类型和相对位置(图2)。描述每个原子局域环境的函数通常被称为描述子(descriptor, 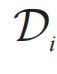 )。描述子一般是高维函数,可以是邻居原子类型和相对位置的直接函数,也可以是定义在原子上的信息,经过多层邻居原子间消息传递(message-passing)之后获得的函数[23]。无论何种情形,某个原子的描述子都是其局域环境的函数。因此,BP能量分解形式的成立依赖于“局域性假设”,也因此保障了能量函数的广延性质。这里需要注意,无论是局域环境到描述子的映射,还是描述子到原子能量贡献Eatom的映射,都是高维非线性函数,体现了原子间相互作用的多体特征,因此特别适合使用深度学习工具进行建模。
)。描述子一般是高维函数,可以是邻居原子类型和相对位置的直接函数,也可以是定义在原子上的信息,经过多层邻居原子间消息传递(message-passing)之后获得的函数[23]。无论何种情形,某个原子的描述子都是其局域环境的函数。因此,BP能量分解形式的成立依赖于“局域性假设”,也因此保障了能量函数的广延性质。这里需要注意,无论是局域环境到描述子的映射,还是描述子到原子能量贡献Eatom的映射,都是高维非线性函数,体现了原子间相互作用的多体特征,因此特别适合使用深度学习工具进行建模。

图2 原子i 局部环境示意图
如果局域性假设成立,我们即可以在相对较小的原子体系上训练原子能量贡献Eatom的模型。进而通过BP分解形式,使用这个模型在任意大的原子体系中计算势能,且计算开销随体系规模线性增长。这带来的好处是非常显然的:我们只需在小体系上进行密度泛函计算产生训练模型的标签,而非在实际应用这个势能的较大体系上。注意密度泛函理论的计算复杂度至少为三次方标度,体系稍大即非常昂贵。当然从较小训练体系到较大应用体系的泛化过程并不是简单直接的。这个泛化问题可以归纳为,训练集小体系上的局域环境是否能够代表任意大的原子体系的局域环境。为了使训练局域环境具有足够代表性,我们需要设计算法使得其覆盖范围尽可能广(详见3.2节的讨论),另外需要系统性增大训练体系确保小体系局域环境不被有限体系效应过度约束。幸运的是,在绝大多数问题中,局域性假设都被很好地满足,一般数十个原子的训练体系就能获得可以泛化到更大体系的势函数模型。
当然,并不是所有的原子间作用都是局域性的,例如库仑相互作用就是本质性长程的相互作用。库仑相互作用在密度泛函理论中是被显式计算的,但是在机器学习势函数中,只能被局域的原子能量贡献等效地描述。对于库仑相互作用不关键的体系,例如存在电荷屏蔽效应的金属体系,局域性假设一般能够成立。在库仑相互作用显著的体系中,较小体系训练的势函数模型无法泛化到任意大体系,为了克服这个困难,显式计算体系中的库仑相互作用成为必须考虑的建模问题。由于势函数输入仅有原子坐标和类型,因此计算库仑相互作用的关键科学问题为如何从原子核坐标和类型信息中近似恢复体系的电子密度,使得由此计算的库仑相互作用具有精度。这里包括了基于部分电荷[24]或瓦尼尔中心[25]的机器学习方法,或者通过数据驱动的方式直接建模长程相互作用[26]的方法。
在建模原子能量贡献的过程中,势能的对称性是一个必须考虑的问题,即我们对所有原子核的坐标施以平移、旋转操作,或者对同种原子核的坐标施以交换操作,势能函数的值保持不变。特别地,作为充分条件,我们要求BP分解中,每个原子能量贡献满足上述对称性。势能函数的对称性要求是符合直觉的:在其他条件不变的前提下,一块材料不会因为从北京移动到了上海就发生了相变。根据Noether定理[27],保守物理系统的每一个连续对称性要求都对应着一个守恒律,这里势函数的平移、旋转对称性对应体系的动量和角动量守恒律。进一步,势函数本身的对称性要求对于训练精度也是至关重要的,图3(a)对比了是否满足交换对称性的模型在训练中的误差下降速度,可见在相同数据和训练步数下,满足对称性要求的模型的精度比不满足的模型高了一个量级以上。此外,原子受力是保守的,这要求其由势函数的负梯度计算得到,并且势函数本身至少是一阶可导的。图3(b)展示了存在不连续性的模型在实际分子模拟中能量发散的情形。
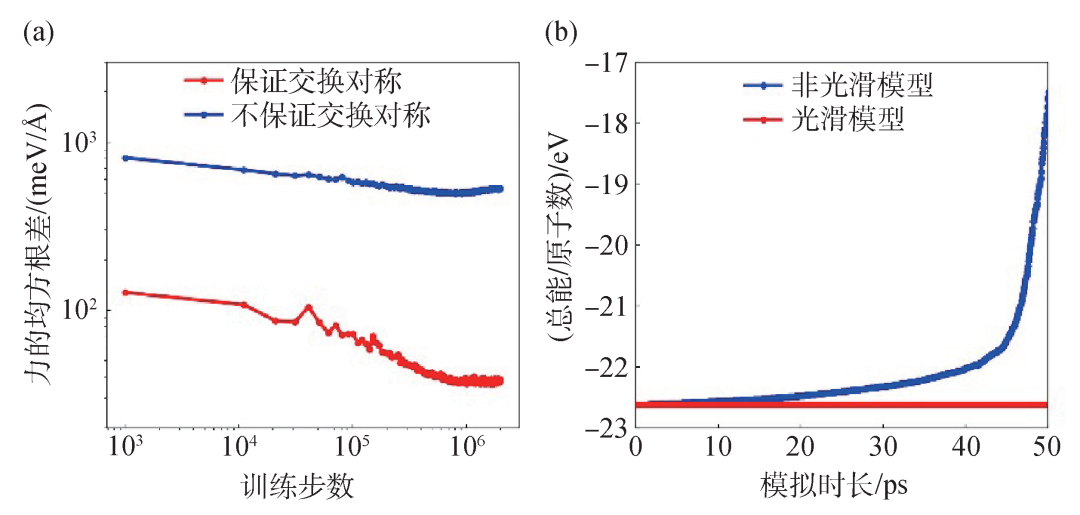
图3 (a)在模型训练中保证交换对称性(红)和不保证交换对称(蓝)模型的学习曲线;(b)在求解牛顿运动方程时,光滑模型(红)和非光滑模型(蓝)的总能随时间变化的情况
机器学习势函数可以以标准的监督学习方式进行训练。由于密度泛函理论一般能够经由Hellmann—Feynman定理[28,29]计算原子受力,在周期性边界条件下能够计算应力张量,因此二者与势能一起作为监督目标训练机器学习势函数,即将损失函数写成能量、受力和应力误差的均方线性组合形式。在数学上,这相当于在逼近一个函数的同时,逼近其对原子核坐标和区域张量的导数,这一般意味着更高的逼近精度,以及更少的过拟合可能。损失函数中,能量、受力和应力误差的前置因子为可调节的超参数。
3.2 同步学习方法中的训练数据生成方法
机器学习模型一般不具有分布外泛化能力,这需要训练数据集以充分覆盖模型在应用中可能遇到的推理场景。在计算机视觉和自然语言处理领域,生成数据的方式一般为图片和语料的收集。理想情况下,只要穷尽已数字化的图像和语料就可以认为是一个充分好的训练数据集,目前计算机视觉和自然语言处理等领域的模型的应用效果已经充分印证了这一点。
在分子模拟领域,高质量模型要求其训练数据的局域原子环境充分覆盖分子模拟中可能遇到的局域原子环境。局域原子环境由描述子刻画,一般而言是高维函数(典型的维数为102量级),如何在高维空间中体现“充分覆盖”并不是一个平凡问题。另一方面,机器学习势函数模型的训练数据一般通过KS密度泛函理论计算获得。对于大多数问题而言这样的数据往往是非常缺乏的,甚至在多数情况下,研究者需要针对特定问题从头生成训练数据。另一方面,与计算机视觉和自然语言处理领域不同,机器学习势函数的训练数据产生代价非常昂贵。这意味着,我们需要使用尽量少的数据,做到在应用问题中对可能遇到的局域原子环境的充分覆盖。
为了达到上述目标,主动学习和同步学习方法被发展出来[30,31],这里我们暂时不讨论这两种方法的细致区别,而统称其为同步学习。这类方法模拟了人类在学校学习的方式:学习——考试查找知识漏洞——学习弥补漏洞——考试查找知识漏洞——学习弥补漏洞……。这个过程可以一直循环,直至所有知识被完全掌握为止。在同步学习方法中,在考试中出卷子对应着通过当下已有模型进行原子局域环境采样;判卷子对应着通过模型误差估计发现误差大的构型;学习弥补漏洞对应着对误差大的构型打标签并重新训练模型。在这个过程中,“试卷”的出题范围必须不小于模型的应用范围,即将来可能用到的知识点必须都被考过。同步学习方法中,仅在最关键的数据点(被判定为模型误差大的数据点)进行KS密度泛函计算,因此在应用范围内达到一致精度的同时,标签计算开销达到近似最小。
相比于从头算分子动力学模拟等标签产生方法,同步学习拥有高得多的数据效率。例如,张林峰等人使用15.3万个从头算分子动力学训练数据[22],训练了适用于0—0.2 GPa,238—330 K热力学区间中4个热力学状态的水的势函数模型。而在另一项工作中[32],他们在模型构造相同的前提下,通过同步学习方法,仅仅产生了约3.1万个训练数据,就训练出了适用于0—50 GPa,0—2400 K的机器学习势函数模型,并在这个热力学区域内绘制了水的相图。
3.3 软件实现与基础设施
开发深度学习势函数模型曾经是非常繁重的工作,因为需要手工实现损失函数对模型参数的导数。这个过程中除了需要实现能量对参数的导数,还包括能量对坐标求导后再对参数求导。深度学习在21世纪第二个十年的蓬勃发展带来了优秀的基础设施,例如TensorFlow[33],PyTorch[34]等开源深度学习框架,这使得开发深度学习势函数模型的门槛大幅度降低。特别地,基于这些框架构建的DeePMD-kit[35],Torch-MD[36]等开源深度学习分子模拟软件,已经成为训练和使用机器学习势函数模型的开箱可用的工具。
传统计算任务(比如密度泛函理论计算)一般由一个软件完成,且消耗的计算资源在软件运行期内较为恒定。作为对比,同步学习在训练和探索阶段需要调用深度学习分子模拟软件,典型的训练任务一般使用少数几个GPU数个小时,而探索任务一般使用数百个GPU数分钟,在标签计算阶段一般使用数千CPU核数小时。这三种任务形成一个循环,整个流程需要复杂的逻辑判断加以控制,以处理循环收敛、进入下一阶段或者因错误停止等不同情况。总结而言,同步学习任务具有工作流程逻辑复杂、调用软件多(依赖冲突可能发生)、算力异质程度高(CPU,GPU计算资源均有调用)、算力需求量弹性极大等特点。手工管理上述工作流程,分配计算资源极度困难且效率低下,而脚本工具可维护性差,难以处理逻辑复杂工作流及进行容错处理。
在这个背景下,dflow[37]等基于argo引擎1)的工作流开发工具被发展出来,支持复杂工作流逻辑,通过容器技术解决不同软件跨平台部署的问题,通过部署云计算资源实现异质算力的高弹性调度,提供丰富的工作流容错及重启动机制。基于dflow[37]开发的一系列工作流也正在被用于各类不同的科学应用中,例如用于合金性质计算的APEX[38]、用于动态催化计算的dynacat-tesla2),等等。这类工具正在成为机器学习势函数训练及应用,以及从更广义的角度而言,机器学习结合科学应用的基础设施之一。
值得关注的是,推动机器学习与人工智能发展的要素中,除核心算法与模型框架外,软件、高性能算力、数据工程等要素和GitHub、Hugging Face等开源社区开放的协同模式是同等重要的。这些要素与模式对于结合机器学习与人工智能的物理建模任务来说同等重要,但常常会被忽略。基于此,我们推荐读者关注DeepModeling开源社区3)和围绕大原子模型主题的OpenLAM4)等实践。

04
挑战与机遇:从“模型”到“大模型”
长期以来,机器学习势函数模型构建遵循着“一事一议”的专用模型建模原则,即针对特定应用问题,通过同步学习方法生成适配的训练数据集,再在该数据集上训练出适用于解决该问题的机器学习势函数模型。沿着这条设计思路开发的机器学习模型已经在许多应用问题中取得了良好的应用效果。特定应用问题的不断累积,针对相互关联的应用问题生成的训练数据往往具有大量公共的信息。例如,针对铝合金设计问题发展的掺杂不同金属元素的势函数训练数据,由于具有同样的铝基底,因此大量局域构型信息是高度类似的。再例如,针对水相图计算产生的训练数据,无论对水还是冰相均有充分好的覆盖,因此稍加扩充即可应用于研究过冷水的结冰过程。因此,在针对一个新的应用问题进行训练时,如果能充分利用已有数据集中的信息,就有望进一步减小新应用问题的训练数据需求,从而大幅节省生成新模型所需的密度泛函理论计算开销。沿着这个思路产生了两种不同的模型训练范式:通用原子模型和大原子模型。
4.1 通用原子模型
通用原子模型将数据信息共享的理念推进至极致:希望针对元素周期表中所有元素产生一个训练数据集,并从该数据集中训练出适用于所有应用问题的通用机器学习势函数模型。这种思路的内在观察是,元素周期表中所有的化学元素原子间相互作用,都由统一的物理模型,即薛定谔方程,或者其近似模型,如密度泛函理论求解获得,因此通用于所有化学元素的势函数模型一定存在。如何获得通用模型可转化为两个问题:(1)怎样的数据集充分代表所有应用中可能遇到的局域原子环境;(2)怎样的模型构造能够在上述数据集上获得良好精度。
通用原子模型要求覆盖元素尽可能充分,并且按照统一的交换关联泛函进行标签计算。目前通用性模型的代表案例,如CHGNet[39]和MACE[40],它们基于材料晶体数据库Materials Project(MP)中构型的结构优化轨迹(MPtrj)训练[39],总数据量约为160万,标签使用的交换关联泛函为PBE[41]或PBE+U[42]。又如,GNoME[43]采用NequIP模型架构[44],在MP数据集基础上,经过6轮针对材料生成的主动学习迭代生成训练数据集,数据集包含约1千万训练数据。该工作声称GNoME模型能量预测的平均绝对误差达到28 meV/atom,可以发现220万种稳定结构,其中38.1万种结构为能量稳定的新材料。上述几个代表性通用模型的训练数据集均针对晶体材料构型的优化获得,因此模型适用于给定材料初始构型的优化工作,例如GNoME应用的材料设计工作流中,需要使用势能函数的步骤即为结构优化。与此同时,上述模型在其他类型的势能函数任务中的精度是缺乏保证的。比如基本的材料力学性质——弹性常数计算及层错能的预测,通用模型MACE的精度相比专用模型尚有较大差距[38]。
通用原子模型要求所有数据采用统一的方式打标签,这使其难以利用不同领域中经过长时间系统积累产生的数据集。例如,化学领域中,ANI数据集包括了约2千万小分子从头算分子动力学模拟生成数据[45],该数据集以ωB97x杂化交换关联泛函[46]打标签。再例如,催化材料领域OC20数据集包含2.6亿催化体系结构优化产生的构型[47],该数据集以RPBE (revised PBE)[48]交换关联打标签。这两个数据集均无法加入通用原子模型训练,其原因是,虽然三个数据集(包括训练通用原子模型的MP数据集)都以密度泛函理论打标签,但是使用的交换关联泛函不同,他们本质上定义了三个不同的势能函数,因此无法放在一起进行训练。因此通用模型如需应用于解决化学或者催化问题,需要重新产生训练数据,或者按照一致的方式重新标签已有数据集。
通用原子模型精度受限于其标签时选定的交换关联泛函的精度。CHGNet,MACE,GNoME均只具有PBE/PBE+U的精度。该精度对于绝大多数材料计算问题是足够的,但是仍然存在大量应用问题需要更高的交换关联泛函精度。例如化学领域一般需要杂化泛函[49,50],对于冰不同相的相对稳定性的正确描述需要SCAN泛函[51]。由此可见,针对不同应用问题的实际需求,泛函的选取需要做到尽量灵活,而通用原子模型一旦训练完成,就失去了这方面的灵活性。
4.2 大原子模型
虽然微尺度研究相关领域很早便有利用神经网络处理大规模数据或表示势函数等物理量的实践,但这些技术直到最近几年才真正爆发并被广泛应用。这得益于算法、数据、算力及软件框架的协同发展。在过去,科研工作者对理论算法的重视程度要远高于其他方面,但技术体系的发展呼唤新的科研模式。类似地,人工智能的发展也得益于良好的算法工程化实践以及开源开放的平台化模式。
在此基础上,正如语言世界中数据的积累推动了机器学习的发展并最终促成了大语言模型(large language model,LLM)的诞生一样,原子世界数据的系统化积累正在让通用大原子模型(large atom model,LAM)的出现成为可能。
不同领域中,大模型的定义不尽相同。一般而言,大模型被认为提供了领域知识的公共表示,其训练精度满足特定标度定律(scaling law)[52],且具有少数帧(few-shot)[53]泛化能力。对于势函数构造领域,由于第3.2节讨论的原因,训练数据量并不直接线性对应于数据中蕴含的信息量,因此尚难以总结标度定律,但是其少数帧泛化能力仍然可以作为评价一个势函数模型是否具有大模型特征的重要参考。
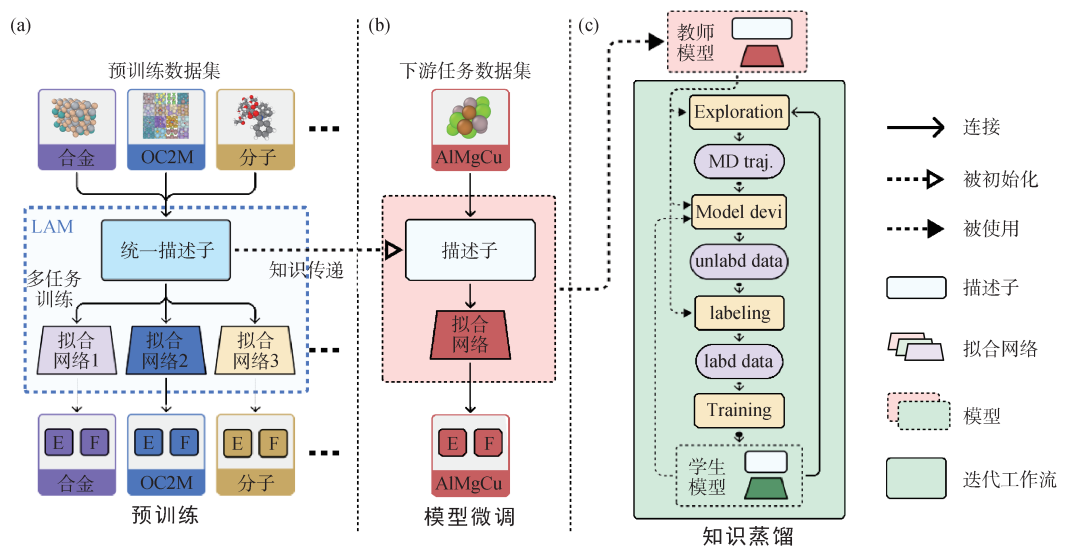
图4 DPA-2大原子模型工作流程示意图 (a)多任务预训练(multi-task pre-training)。不同DFT标签的数据可以通过共享单个描述子并拥有各自的拟合网络来一起预训练,这让我们能够得到一个统一的描述子(unified descriptor);(b)在下游数据集(down‐stream dataset)上进行模型微调(fine-tuning);(c)蒸馏(distillation)过程。使用微调模型作为教师模型,迭代执行分子动力学(MD)模拟,并将标记数据添加到训练集中,以训练一个高效率的学生模型,便于下游应用[54]
大原子模型的一个实现,DPA-2 (deep potential attention-2)[54]的模型工作流程如图4所示。其借鉴了自然语言处理等领域大模型的成功经验,将工作流分成了预训练—下游微调—知识蒸馏三个步骤进行。
获得少数帧泛化能力的关键是在预训练阶段以尽可能多的、来源于不同领域的数据集(包括已有数据集和为大模型生成的数据集)进行训练,在预训练模型中凝练不同数据集中化学知识和原子构型知识的公共表示。这是大原子模型区别于通用原子模型的最显著特征。例如,在体相材料数据集以及化学分子数据集中获得的知识,有利于在催化体系中提升模型的泛化能力(这里催化体系指,将小分子放置在金属或氧化物体相材料的自由表面形成的体系)。为了解决不同数据集间密度泛函理论计算标签不统一的问题,DPA-2采用了多任务训练(multi-task training)的训练模式。首先描述子部分为模型的公共结构,在其上连接出若干个互相独立的拟合网络,每个拟合网络针对一个数据集进行训练。DPA-2设计了具有强表示能力的描述子,具体而言,为基于消息传递机制的多层表示进化网络。DPA-2同时弱化拟合网络的表达能力,特别地,取每个拟合网络为标准的前馈全连接网络。在这样的网络架构下,不同数据集间的公共知识可以被凝练于公共的描述子表示中,而不同交换关联泛函带来的标签特化信息被拟合网络所捕捉。经过预训练的描述子和拟合网络即为大原子模型。
在下游任务中,描述子得以保留,拟合网络选取与下游任务类似的网络,或者进行随机初始化。DPA-2在全部15个下游任务中均展现了少数帧泛化能力:相比白板模型,预训练模型可以使用少1—3个量级的数据达到同样训练精度,这意味着下游任务需要进行的密度泛函标签计算开销减小了1—3个量级。
由于需要提取大量数据集中的公共知识,DPA-2需要使用表达能力非常强的描述子提取数据中的公共知识表示,但这造成了下游任务模型计算开销比较昂贵。同时,对于特定下游任务,由于其原子构型和元素的丰富程度远不及预训练集,也无需使用表达能力过强的模型结构。因此在完成下游微调后,DPA-2使用模型知识蒸馏技术,将下游模型蒸馏至一个具有高运行效率的专有模型。特别地,蒸馏过程使用下游微调模型作为教师模型生成训练标签,在同步学习循环中训练专有的学生模型。知识蒸馏可以在模型精度几乎不受损的前提下,提升模拟效率高达2个数量级,蒸馏后模型的运行精度和专有模型完全一致。
4.3 大原子模型在其他任务中的潜在机会
大原子模型的发展既是一个正在涌现的机遇,也仍面临很多挑战。有了它能做什么?我们期待它能在多类任务中展示其出色的迁移和泛化能力。首先,我们已经介绍,期待它迁移到势函数等关键物理量的表示上,避免了生成大量昂贵的量子力学数据的必要性,从而直接解决实际问题。其次,大原子模型应能更有效地拟合实验性质、充分利用小样本数据,从而实现更精准的构效关系建模。此外,大原子模型的生成能力,应能系统性地革新现有的结构搜索和增强采样算法。最后,我们期望该模型能根据特定需求优化结构设计,如依据药效和安全性要求直接设计药物分子,或按所需物理化学性质设计材料,并能更高效准确地解析实验表征信号、推荐实验设计参数,在原子级的生产制造任务中发挥“智能大脑”的作用。这些进步预计将极大推动科学研究和工业应用的革新。

05
总结与展望
本文整体介绍了从薛定谔方程到大原子模型的发展历程,特别关注了近十年来机器学习在原子尺度模拟中的应用与进展。文章首先回顾了微观模拟的基本原理和数值方法,随后深入探讨了机器学习与物理结合的建模过程,并强调了数据积累及软件工程基础设施在该领域发展中的关键作用。最后,文章介绍了大原子模型的最新进展,并对其在多个科学领域中的潜在应用进行了广泛讨论。
大原子模型作为一个迅速发展的研究领域,面临着诸多挑战与机遇。我们预期模型的泛化、迁移和生成能力能在可见的未来迅速提升,也期待大原子模型与原子级实验表征及生产制造实现更深层次的融合。此外,构建更大规模和更多样化的数据集,以及开发更先进的软件和工作流管理工具,对于推动该领域的发展至关重要;跨学科应用的深入探索和开源协作的广泛推进,将为大原子模型的实际应用开辟更广阔的前景。
注:
1) https://argoproj.github.io
2) https://app.bohrium.dp.tech/dynacat-md/
3) https://github.com/deepmodeling
4) https://deepmodeling.com/blog/openlam/
参考文献
[1] Feynman R P. The Feynman Lectures on Physics. http://www.feynman lectures.caltech.edu
[2] Dirac P A M. Proceedings of the Royal Society of London,series A,1929,123(792):714
[3] Schrödinger E. Physical Review,1926,28(6):1049
[4] Kohn W. Reviews of Modern Physics,1999,71(5):1253
[5] Pauli W. Exclusion Principle and Quantum Mechanics. In:Writings on Physics and Philosophy,Springer,1946. pp.165—181
[6] Hartree D R. The Wave Mechanics of an Atom with a Noncoulomb Central Field. Part I. Theory and Methods. In:Mathematical Proceedings of the Cambridge Philosophical Society,Cambridge University Press,1928. pp.89—110
[7] Fock V. Zeitschrift für Physik,1930,61:126
[8] Hylleraas E A. Zeitschrift für Physik,1928,48(7):469
[9] Čížek J. The Journal of Chemical Physics,1966,45(11):4256
[10] Ceperley D,Chester G V,Kalos M H. Physical Review B,1977,16(7):3081
[11] White S R. Phys. Rev. Lett.,1992,69(19):2863
[12] Knizia G,Chan G K L. Phys. Rev. Lett.,2012,109(18):186404
[13] Han J Q,Zhang L F et al. Physics Today,2021,74(7):36
[14] Born M,Oppenheimer R. Annalen der Physik,1927,389(20):457
[15] Hohenberg P,Kohn W. Physical Review,1964,136(3B):B864
[16] Kohn W,Sham L J. Physical Review,1965,140(4A):A1133
[17] Medvedev M G,Bushmarinov I S,Sun J W et al. Science,2017,355(6320):49
[18] Car R,Parrinello M. Phys. Rev. Lett.,1985,55(22):2471
[19] Jia W L,Wang H,Chen M H et al. Pushing the Limit of Molecular Dynamics With ab initio Accuracy to 100 Million Atoms with Machine Learning. In:SC20:International Conference for High Performance Computing,Networking,Storage and Analysis,IEEE,2020.pp.1—14
[20] Jumper J,Evans R,Pritzel A et al. Nature,2021,596(7873):583
[21] Behler J,Parrinello M. Phys. Rev. Lett.,2007,98(14):146401
[22] Zhang L F,Han J W,Wang H et al. Phys. Rev. Lett.,2018,120(14):143001
[23] Schütt K,Kindermans P J,Felix H E S et al. SchNet:A Continuous-filter Convolutional Neural Network for Modeling Quantum Interactions. In:Advances in Neural Information Processing Systems 30 (NIPS 2017)
[24] Artrith N,Morawietz T,Behler J. Physical Review B,2011,83(15):153101
[25] Zhang L F,Wang H,Muniz M C et al. The Journal of Chemical Physics,2022,156(12):124107
[26] Grisafi A,Ceriotti M. The Journal of Chemical Physics,2019,151(20):204105
[27] Noether E. Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen,Mathematisch-Physikalische Klasse,1918,1918:235
[28] Hellmann H. Einfuhrung in die quantenchemie. Springer,1937
[29] Feynman R P. Physical Review,1939,56(4):340
[30] Smith J S,Nebgen B,Lubbers N et al. The Journal of Chemical Physics,2018,148(24):241733
[31] Zhang L F,Lin D Y,Wang H et al. Physical Review Materials,2019,3(2):023804
[32] Zhang L F,Wang H,Car R et al. Phys. Rev. Lett.,2021,126(23):236001
[33] Abadi M,Barham P,Chen J M et al. TensorFlow:A System for Large-Scale Machine Learning. In:12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16),2016. pp.265—283
[34] Paszke A,Gross S,Massa F et al. Advances in Neural Information Processing Systems,2019,32:8026
[35] Wang H,Zhang L F,Han J Q et al. Computer Physics Communications,2018,228:178
[36] Doerr S,Majewski M,Pérez A et al. Journal of Chemical Theory and Computation,2021,17(4):2355
[37] Liu X,Han Y B,Li Z Y et al. Dflow:A Python Framework for Constructing Cloud-native Ai-for-science Workflows. 2024,arXiv:2404.18392
[38] Li Z Y,Wen T Q,Zhang Y Z et al. An Extendable Cloud-native Alloy Property Explorer. 2024,arXiv:2404.17330
[39] Deng B W,Zhong P C,Jun K et al. Nature Machine Intelligence,2023,5(9):1031
[40] Batatia I,Benner P,Chiang Y et al. A Foundation Model ForAtomistic Materials Chemistry. 2023,arXiv:2401.00096
[41] Perdew J P,Burke K,Ernzerhof M. Phys. Rev. Lett.,1996,77(18):3865
[42] Anisimov V I,Zaanen J,Andersen O K. Physical Review B,1991,44(3):943
[43] Merchant A,Batzner S,Schoenholz S S et al. Nature,2023,624:80
[44] Batzner S,Musaelian A,Sun L X et al. SE (3)-equivariant Graph Neural Networks For Data-Efficient And Accurate Interatomic Potentials. 2021,arXiv:2101.03164
[45] Smith J S,Isayev O,Roitberg A E. Scientific Data,2017,4(1):1
[46] Chai J D,Head-Gordon M. Physical Chemistry Chemical Physics,2008,10(44):6615
[47] Chanussot L,Das A,Goyal S et al. ACS Catalysis,2021,11(10):6059
[48] Hammer B,Hansen L B,Nørskov J K. Physical Review B,1999,59(11):7413
[49] Becke A D. The Journal of Chemical Physics,1993,98(2):1372
[50] Mardirossian N,Head-Gordon M. Molecular Physics,2017,115(19):2315
[51] Sun J W,Ruzsinszky A,Perdew J P. Phys. Rev. Lett.,2015,115(3):036402
[52] Kaplan J,McCandlish S,Henighan T et al. Scaling Laws for Neural Language Models. 2020,arXiv:2001.08361
[53] Brown T,Mann B,Ryder N et al. Advances in Neural Information Processing Systems,2020,33:1877
[54] Zhang D,Liu X,Zhang X Y et al. DPA-2:Towards a Universal Large Atomic Model for Molecular and Material Simulation.2023,arXiv:2312.15492
(参考文献可上下滑动查看)
//
《物理》50年精选文章
我对吴有训、叶企孙、萨本栋先生的点滴回忆 | 《物理》50年精选文章
国立西南联合大学物理系——抗日战争时期中国物理学界的一支奇葩(Ⅰ) | 《物理》50年精选文章
国立西南联合大学物理系——抗日战争时期中国物理学界的一支奇葩(Ⅱ) | 《物理》50年精选文章
原子核裂变的发现:历史与教训——纪念原子核裂变现象发现60周年 | 《物理》50年精选文章
回顾与展望——纪念量子论诞生100周年 | 《物理》50年精选文章
中国理论物理学家与生物学家结合的典范——回顾汤佩松和王竹溪先生对植物细胞水分关系研究的历史性贡献(上) |《物理》50年精选文章
中国理论物理学家与生物学家结合的典范——回顾汤佩松和王竹溪先生对植物细胞水分关系研究的历史性贡献(下) |《物理》50年精选文章
为了忘却的怀念——回忆晚年的叶企孙 | 《物理》50年精选文章
从分子生物学的历程看学科交叉——纪念金螺旋论文发表50周年 | 《物理》50年精选文章
美丽是可以表述的——描述花卉形态的数理方程 | 《物理》50年精选文章
一本培养了几代物理学家的经典著作 ——评《晶格动力学理论》 |《物理》50年精选文章
熵非商——the Myth of Entropy |《物理》50年精选文章
普渡琐记——从2010年诺贝尔化学奖谈起 |《物理》50年精选文章
天气预报——由经验到物理数学理论和超级计算 | 《物理》50年精选文章
纪念Bohr的《伟大的三部曲》发表100周年暨北京大学物理专业建系100周年 | 《物理》50年精选文章
凝聚态材料中的拓扑相与拓扑相变——2016年诺贝尔物理学奖解读 |《物理》50年精选文章
通用量子计算机和容错量子计算——概念、现状和展望 | 《物理》50年精选文章
谈书说人之一:《理论物理学教程》是怎样写成的?| 《物理》50年精选文章
时空奇点和黑洞 ——2020年诺贝尔物理学奖解读 |《物理》50年精选文章
凝聚态物理学的新篇章——超越朗道范式的拓扑量子物态 | 《物理》50年精选文章
对于麦克斯韦方程组,洛伦兹变换的低速极限是伽利略变换吗?| 《物理》50年精选文章
杨振宁先生的研究品味和风格及其对培育杰出人才的启示 | 《物理》50年精选文章
庞加莱的狭义相对论之一:洛伦兹群的发现 | 《物理》50年精选文章



















 1254
1254

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









