






 文章探讨了预测化学在逆合成路径设计、反应条件优化和新反应发现中的应用。模板-based和template-free方法用于一步和多步逆合成预测,机器学习模型在反应类型分类和产物预测中展现出优势。此外,强化学习被提出来自动化发现反应机制。尽管取得进展,但模型的解释性和外推能力仍待提高。
文章探讨了预测化学在逆合成路径设计、反应条件优化和新反应发现中的应用。模板-based和template-free方法用于一步和多步逆合成预测,机器学习模型在反应类型分类和产物预测中展现出优势。此外,强化学习被提出来自动化发现反应机制。尽管取得进展,但模型的解释性和外推能力仍待提高。
文章概述
- 关键词:预测化学,逆合成,有机反应发现,前沿合成化学
- 内容:预测化学与描述分子如何交互和反应的模型有关,文章总结了目前预测化学相关技术和发展趋势
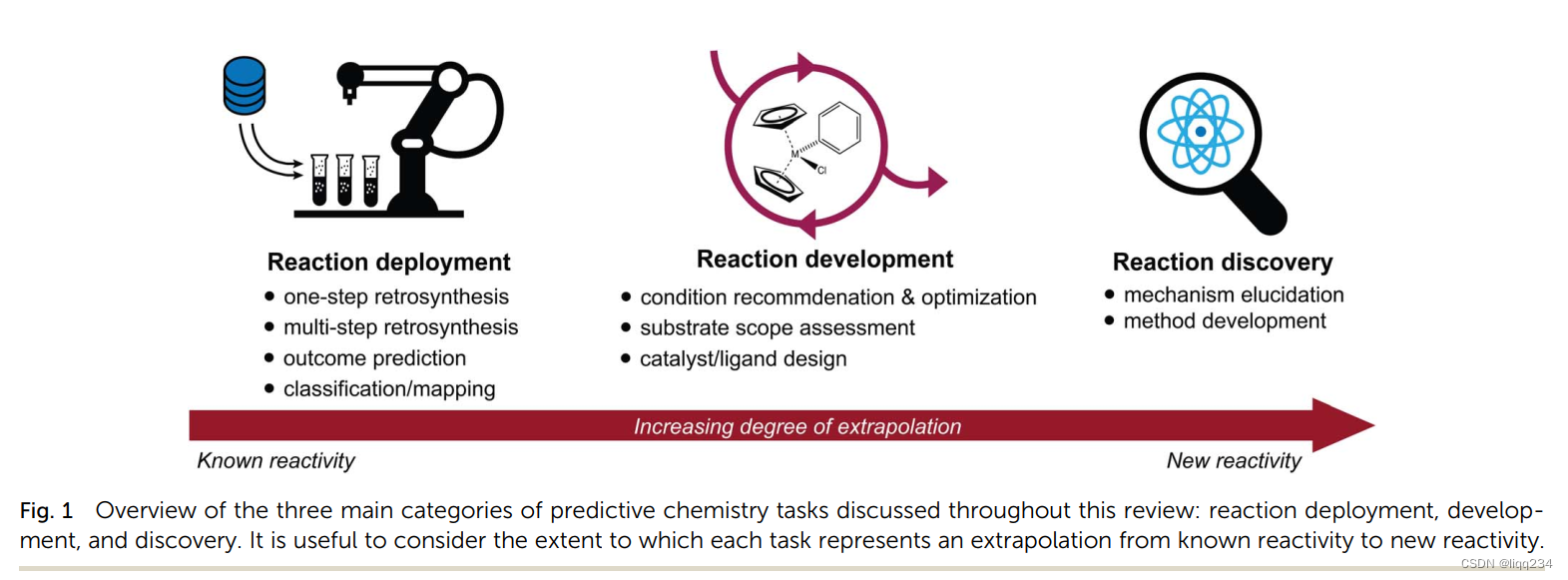
文章内容
Reaction deployment
- 概念:从反应语料中学习趋势和预测已知的反应何时可用于新型反应底物和相互结合。
- 目标:基于对反应数据的算法分析或者统计学分析进行逆合成路径设计和反应预测
- One-step retrosynthetic prediction: 给定产物分子预测“正确”的反应前驱体。根据方法是否使用reaction templates, 分为template-based 方法和template-free 方法;template-free方法又可分为graph-edit方法和translation-based 方法
- Template-based方法:可有人工设计或者从反应数据中提取出反应物和产物之间的结构转换规则。可以使用RDKit工具由产物推出相关的反应物,或者使用RDChiral提取templates。除了分类的方法,还可以使用将一步逆合成看作是检索或者排序问题
- Template-free graph-edit based approaches:将目标产物的分子图转换为反应物分子图。而图编辑方法的变体-基于半模板的方法近期得到广泛关注,首先将产物的原子和键断成假设的中间体,通过这些中间体将其恢复成可能的反应物。反应物恢复的过程可建模为leaving group selection, graph generation以及sequence generation。
- Template-free translation-based approaches:这种方法不需要做atom-mapping,使得模型更容易训练。
- Reranking, transfer learning and retrieval-based methods:使用对比学习方法训练重排模型。迁移学习可以将从大型语料中学到的预训练模型应用到更小的数据集中。基于检索的方法可以直接从已有的分子中找到可能的产物,但是这种方法限制了模型的泛化能力,因为它假设了数据集合包含了反应物所有可能的结构。
- Multi-step retrosynthetic planning:多步逆合成设计可以分为三个阶段-选择、扩展、更新;一个关键的挑战是评估筛选出来的路径是好与坏。
- Monte Carlo tree search (MCTS) for multi-step planning:使用MCTS找出可能的路径并在搜索的过程中用一个分类器迅速查看当前反应是否合理。
- Improvement of the search algorithm and structure:可以将搜索树转换为multi-tree,或者使用一些替代的方法例如Proof Number Search(PNS)来进行搜索。
- Improvement of the selection policy:在整个搜索中,使用一些启发式方法对反应进行评估。
- Enumeration, ranking and clustering of pathways:使用不同的方法对多条路径进行搜索,排序,得到多个可行路径。
- Retrosynthesis-derived models for synthetic complexity:使用逆合成设计工具评测合成的复杂性。
- Reaction outcome prediction:使用一些计算化学的方法预测反应的产物。
- Template-based and template-free major product prediction:1-我们可以将正向预测建模为反应类型分类或模板分类,类似于一步反合成的基于模板的方法。给定一组反应物,目标是预测反应的类型,这隐含地确定了一种或多种产物。Coley等人后来提出了一种两阶段的变体来预测产物分子本身,其中预提取的大约1700个模板被详尽地应用于任何反应物以生成候选产物列表,然后通过学习反应似然估计器对其重新排序以产生最终建议。2-基于图形编辑或基于翻译,唯一基于模板的竞争对手是localtransform,它适应了更通用的反应模板定义。最值得注意的是,基于翻译的模型,如molecular transformer和后续模型,在重现实验观察到的反应产物的准确性方面,在基准数据集(如USPTO_480k)上显示出明显优于其他方法的优势。
- 将预测建模为反应类型分类或者模板分类的方法,根据反应的类型可以隐含的推测出可能的产物。
- Coley等人后来提出了一种两阶段的变体来预测产物分子本身,其中预提取的大约1700个模板被详尽地应用于任何反应物以生成候选产物列表,然后通过学习反应似然估计器对其重新排序以产生最终建议。
- 免模板方法:图编辑方法可分为两阶段的pipeline和顺序图形编辑公式。用于反应预测的两阶段公式与用于反合成的公式类似,实际上比它们早了很多年。与反合成的主要区别在于,反合成的反应中心是跨越多个反应物分子的10个原子对,而不是来自单一的目标产物;正如我们在反合成部分所讨论的那样,在MEGAN中提出的顺序石墨烯配方也可以很好地用于反应结果预测-通过反转图形编辑序列。最后但并非最不重要的是,使用qm增强图神经网络可以作为化学直觉的一种形式,因为基于结构和基于描述符的表示的组合在类似背景下的样本外预测上取得了有希望的结果。
- 免模板方法:另一方面,将基于翻译的方法用于反应预测是相当直接的;它仍然是smile到smiles的转换,只是现在输入和输出交换了。事实上,这些方法的发展几乎与反合成方法的发展趋势完全相同,从基于rnn的序列模型演变为基于变压器的分子变压器,然后使用图形感知编码器,包括GRAT和Graph2SMILES.在反合成部分讨论的一些模型架构和技术也直接应用于正向预测,证实了预训练等技术在正向预测方面的有效性。
- Selectivity prediction for specific reaction types
- Reaction classification and mapping
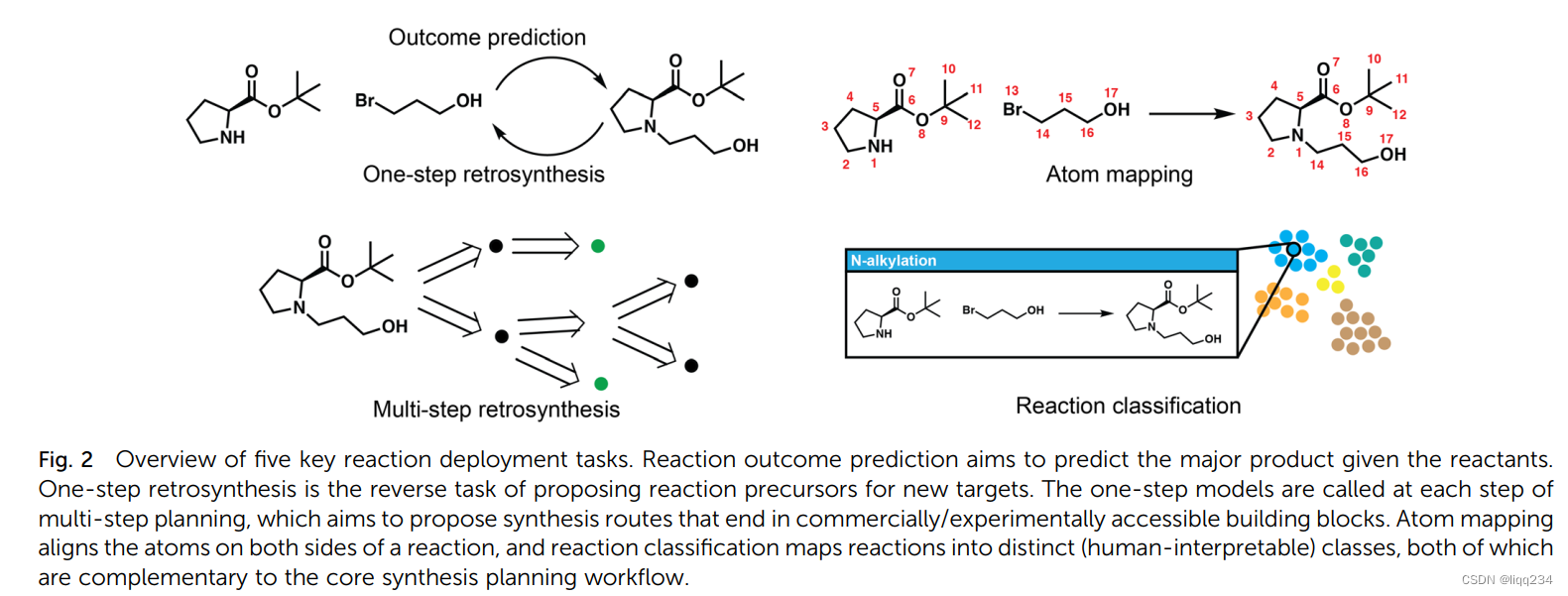
- Template-based and template-free major product prediction:1-我们可以将正向预测建模为反应类型分类或模板分类,类似于一步反合成的基于模板的方法。给定一组反应物,目标是预测反应的类型,这隐含地确定了一种或多种产物。Coley等人后来提出了一种两阶段的变体来预测产物分子本身,其中预提取的大约1700个模板被详尽地应用于任何反应物以生成候选产物列表,然后通过学习反应似然估计器对其重新排序以产生最终建议。2-基于图形编辑或基于翻译,唯一基于模板的竞争对手是localtransform,它适应了更通用的反应模板定义。最值得注意的是,基于翻译的模型,如molecular transformer和后续模型,在重现实验观察到的反应产物的准确性方面,在基准数据集(如USPTO_480k)上显示出明显优于其他方法的优势。
- One-step retrosynthetic prediction: 给定产物分子预测“正确”的反应前驱体。根据方法是否使用reaction templates, 分为template-based 方法和template-free 方法;template-free方法又可分为graph-edit方法和translation-based 方法
Reaction Development
- 概念:加速已存在的化学过程的提升和优化,通常与实验相结合不断迭代循环
- 目标:
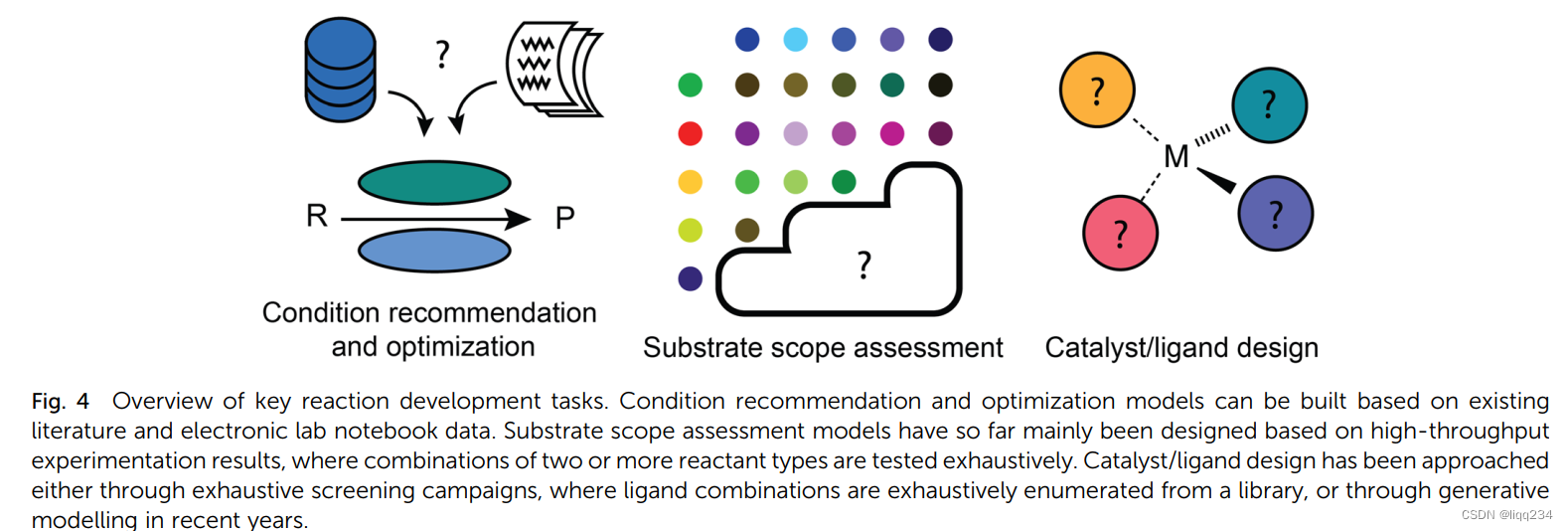
- Reaction condition recommendation and optimization: 与逆合成相关,目前很少有工作探究反应条件的预测。1-最容易的方法是预想一个专家系统在特定的反应族中给出一些定性的推荐,因为对于具体的反应族,很容易给出必要的反应条件。2-而根据可行性,更有用的方法是根据特定底物,依据实际使用的反应物来调整条件。3-Global models与上述模型相反,可以对任何有机反应预测合适的反应条件。Maser等人证明,基于关系图卷积神经网络的单一模型架构可以恢复文献报道的铃木耦合、C-N耦合、根石耦合和Paal-Knorr反应的条件,其精度远远超过仅预测最流行条件的基线方法。缺陷-上述方法都不能准确的给出反应条件,忽略了浓度,添加的顺序等细节,主要的原因是训练数据中缺乏这些信息。解决-目前有两种解决办法,一种是手动构建或生成包含这些定量信息的数据集,或者使用像ORD一样的数据标准,或者使用集中实验的局部模型,其中条件的大多数方面保持不变;第二种是将模型预测作为后续优化活动的初始猜想。
- Substrate scope assessment:合成方法学论文的一个典型部分是底物范围表,展示了反应转换可行时的反应物的广度。这种方法对于计算算法,逆合成设计都很有用,它可以了解提出的反应步骤是否可行。
- Catalyst/ligand design: 1-丹麦和同事使用支持向量机来预测手性磷酸基催化剂的选择性,并为催化剂的选择提供信息;2-Corminboeuf和同事已经应用核脊回归模型来筛选适合均相催化的过渡金属配合物,例如C-C交叉偶联和芳醚裂解反应;具体来说,替代模型预测了特定催化剂沿着所谓的“分子火山图”的相对位置:靠近火山高原的催化剂可以预期表现出理想的底物-催化剂结合特性,从而获得最佳的热力学/动力学过程。除了能够详尽筛选假设的催化剂/配体结构的替代模型之外,生成式ML模型也被开发用于提出新的结构。生成设计本身是一项已有数十年历史的技术,但采用现代ML技术的深度生成设计重新引起了人们的兴趣。目前3-遗传算法可用与均相催化剂优化,遗传算法比起使用原子表示,片段表示,token表示的深度生成模型更能具有创造性的探索正大的设计空间。目前已发布的催化剂/配体数据集:Kraken,OSCAR,Open catalyst Dataset。
Reaction discovery
- 概念:通过对反应机理的解释创造出新的知识并且发现新的物质合成方法
- 发展:1-机器学习算法出现以前,发现通常来自于意外或者通过算法的详尽筛选过程而得到。有关算法/自动化加速化学发现,Gromski等人做了一部分回顾。目前,文章关注于两个方面的内容-促进未知反应机制的解释和新方法/反应开发。
- 目标:
- Elucidation of unknown mechanisms:1-大多数应用于化学反应性的机器学习算法都是机制无关的,也就是说,它们提供给定一组输入的预测结果,但不提供有关化学转化实际如何发生的信息(反应机理的典型解释采用箭头推进图和/或催化循环的形式。然而,有时可以间接地从机器学习分析中获得机械线索)。例如,在他们对pd催化的C-N交叉偶联反应的研究中,Ahnemanet等人根据在他们构建的随机森林模型中从描述符重要性分析中获得的机制线索,确定了一种新的催化剂抑制机制。将预测模型提炼成可解释的决策树,即使模型本身不是固有的可解释,也可以提供洞察力,正如Raccuglia等人所做的那样,他们基于支持向量模型(SVM)推导出决策树,以预测模板化亚硒酸钒的晶体形成。在缺乏先验知识的情况下(随后是仔细的特征工程),这并不一定得到保证,因此这些方法在生成机制理解方面的成功部分取决于意外发现(尽管成功的几率可以通过铸造一个广泛/多样化的输入描述符/模型特征的网络来增加)。2-另一种更系统的方法是使用反应网络,列举+评估得到新的反应机制解释。一种有前景的勘探策略包括反应分子动力学(MD)模拟,根据预先设计的热力学集合对可访问的构型进行采样,例如由Mart ’ ınez及其同事开发的“从头开始纳米反应器”。但是这种方法的计算成本特别高,从而在应用推广上收到了限制。3-其他探索方法采用静态量子化学计算来估计与基本反应步骤相关的过渡态结构和势垒高度。最后,Reiher及其同事的CHEMOTON项目代表了一种通用的、独立于系统的探索方法,该方法基于直接从(静态)电子结构中导出的启发式规则以有效和公正的方式探索复杂的反应网络。但是这种方法具有组合爆炸的风险,可以使用机器学习方法来区分高和低倾向的组合/路径。3-最近,一些小组已经开始采用强化学习技术,以自动化和有效的方式发现机制。强化学习在反应网络探索的背景下具有特殊的前景,因为它绕过了明确枚举和评估基本反应步骤的所有组合的需要,因此,当涉及到反应时,当足够精确时它可以被认为是效率的最终缩影网络探索算法。但是这种方法并不能避免使用第一性原理的计算过程。
- New method development:原则上,传统的反合成和反应预测模型能够提出可以被认为是新颖的转变。在最简单的情况下,无模板的反合成模型可以提出与训练集中不存在的模板相匹配的反应。然而,在实践中,外推的程度往往是有限的。由反应预测模型提出的“新”反应可能涉及对已知模板的微小修改,而底物仅略有改变。例如,Segler和Waller将化学反应空间建模为一个图,其中分子由节点表示,反应由边表示,并应用网络分析技术来预测图中新的可信链接。通过对连接相似分子的网络边缘进行更详细的分析,他们甚至能够为高通量反应发现活动提出有希望的起点。这里应该指出的是,将新反应定义为已知半反应的前所未有的组合可能不是所有化学家都同意的。强力筛选反应物和条件组合至少是一种基本方法。
展望
到目前为止,机器学习辅助机制阐明中的大部分活动都位于描述符重要性策略中。我们希望这些模型能够在不受人类专家指导的情况下产生新的见解,并最终能够产生开放式的假设和发现。为了实现这一目标,我们自己正在进行的预测化学工作的特点是两个过渡:从定性到定量,从回顾性到前瞻性。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









