







 文章提出了一种基于知识图谱的化学推理模型,该模型将反应预测转化为图谱中的链接预测问题,通过寻找反应物分子间的可行路径来预测反应。这种方法强调了在路径的有效性和化学意义上的过滤器,并且可以预测产物和反应条件。研究主要集中在二元反应上,对于未来多元反应的预测具有指导意义。
文章提出了一种基于知识图谱的化学推理模型,该模型将反应预测转化为图谱中的链接预测问题,通过寻找反应物分子间的可行路径来预测反应。这种方法强调了在路径的有效性和化学意义上的过滤器,并且可以预测产物和反应条件。研究主要集中在二元反应上,对于未来多元反应的预测具有指导意义。
文章概述
- 关键词:化学推理,反应预测,反应发现,有机反应,链接预测,知识图谱
- 内容:有机化学家使用构建的知识图谱用于解决合成问题和发明新的转换,文章提出一个模型可以模拟化学推理,将反应预测问题刻画为找到知识图谱中缺失链接的问题。这个模型仅需要依靠图内部结构推理出关于reactivity和reactions的假设,可被作为反应发现的高通量假设生成器。
研究背景
预测反应的要素:反应物的反应性、反应机理
- 因:基于知识图谱的推理预测能力使得有机合成与新转换的搜索变得可能,预测反应物的反应性对于预测反应至关重要。然而,反应性的预测受限于一系列条件,包括反应物,催化剂,因此,反应预测在广义层面上包括在给定反应物的条件下预测产物,催化剂,试剂。HTRP(high-throughput computer-based reaction prediction)可被用于药物设计,虚拟化学空间探索,可合成性评估,或是预测逆合成系统中断开是否可行。
- 发展:HTRP没有被广泛使用是由于巨大的计算开销,因此高通量的反应预测是使用一个模型的系统。这些预测使用基于规则的专家系统,机器学习,形式逻辑以及力场,半经验的结合来实施。1-基于高通量的HTRP方法在其应用的领域表现良好,但是目前在规模上有限并且还不能看到这种方法是否能用在新的反应类型当中,特别是对于转换金属催化反应。2-形式逻辑方法将分子和电子转换描述为矩阵,并将其运用到发现新的周环反应中,但是对于金属转换化学,用这些方法实现预测是最困难的。预测反应条件的工作很少,一般都是预测特定的反应类型。3-Baldi and co-workers 设计了在反应机理层预测反应的方法,不包含金属转换催化过程,然而尽管反应机理对于反应理解至关重要,但是一些反应能被预测是因为他们的完全转换。许多反应的细节性机理是不可知的,并且可靠的数据无法获取。再者,反应机理会随着条件的改变而改变。
- 缺点:专家系统基于手动输入或者算法提取反应规则,是预测反应最广泛使用的方法。但是这种方法有许多缺陷:1.他们需要专家将知识手动编码而不是直接作为反应规则使用或者使用启发式方法从数据中提取规则;2.维护这样的系统非常困难;3.这些规则并不能泛化。专家系统只能使用已有规则预测用这些规则编码的反应,而不能发现新的化学信息。
- 果:因此文章引入了一种新的预测反应的方法,假设预测一个没有见过的反应,忽略它的反应类型,必须在当前收集到的知识图谱中找到反应物分子缺失的链接。
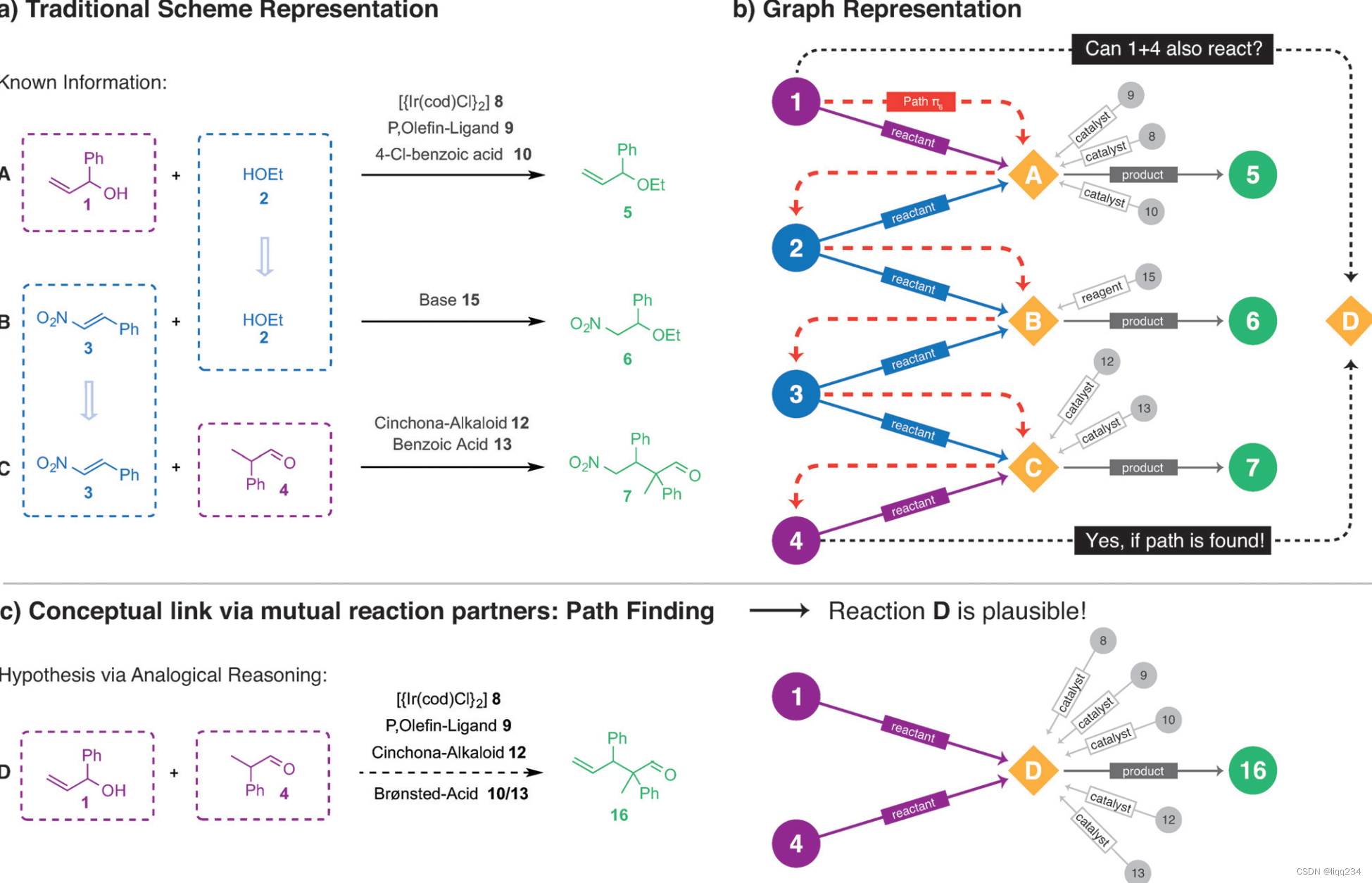
研究方法
知识表示
- 反应分子图:将一个反应表示为图的形式,如上图1,2,3,4等数字表示化合物,他们的角色在图中的边上表示当前化合物与反应的关系。
反应预测看作知识图谱中的链接预测问题
-
找到当前需要预测反应的反应物分子间的可行路径:

-
将反应预测问题当成缺失节点和边预测问题:通过分析路径来预测缺失的链接。
找到 π L = ( m 1 , m 2 , . . . , m n ) \pi^{L} = (m_{1}, m_{2}, ..., m_{n}) πL=(m1,m2,...,mn), 使用如下公式证明其中的分子 m 1 m_{1} m1, m n m_{n} mn是相似并且相互补充的,如果不能找到反应物之间的路径,则不能简单的对反应物之间的反应性做出任何描述:
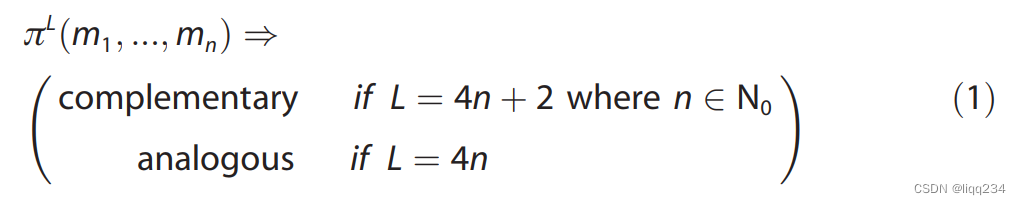
- 确保找到的路径是具有化学意义的,文章定义了两个过滤器:
- 过滤器1:在一条路径上的反应中,前一个反应的 m i m_{i} mi在执行 r Q r_{Q} rQ后原子发生了改变,这条路径才是有效的。
- 过滤器2:一条路径中的反应节点 r J r_{J} rJ和 r J + 1 r_{J+1} rJ+1需要有相同的反应中心,文章使用反应指纹来对反应进行编码。
- 确保找到的路径是具有化学意义的,文章定义了两个过滤器:
-
化学推理算法: 在寻找可行路径的过程中,算法计算反应节点的反应指纹得到Tanimoto相似性,如果相似性低于模型参数例如0.2,则排除当前路径。整个算法使用宽度优先搜索形式直到找到所有的可行路径。

-
产物预测:使用half-reactions的概念,一个二元反应可以分成两个半反应,一个半反应包含反应物的原子和键在产物中如何转换和合并。依靠结合两个半反应的结果,即可得到最终的产物。
-
条件预测:将第一个反应的条件和最后一个反应的条件取并集得到一个初始的反应条件。
-
数据:来自Reaxys的82万二元反应
-
验证方法:time-split验证,使用第一时间之前的所有数据预测这个时间之后发布的反应。
小结
- 对于催化剂,反应类型,反应机理,反应性不用太多关注,只关注于反应图本身获取到可能的产物,反应条件只需要一个简单的并集便可解决。
- 但是目前的模型只针对于二元反应,但是对于多元反应具有很好的参考意义。

















 705
705

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









